

Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice
- PMID: 24591593
- PMCID: PMC3964055
- DOI: 10.1073/pnas.1401595111
Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice
Abstract
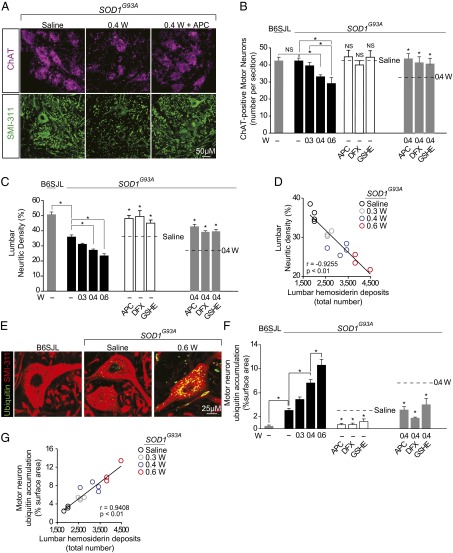
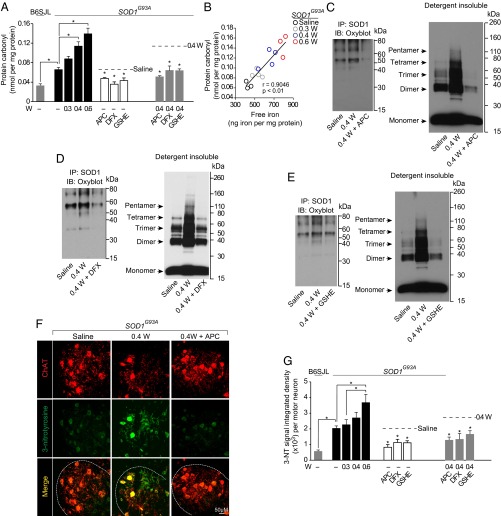
Humans with ALS and transgenic rodents expressing ALS-associated superoxide dismutase (SOD1) mutations develop spontaneous blood-spinal cord barrier (BSCB) breakdown, causing microvascular spinal-cord lesions. The role of BSCB breakdown in ALS disease pathogenesis in humans and mice remains, however, unclear, although chronic blood-brain barrier opening has been shown to facilitate accumulation of toxic blood-derived products in the central nervous system, resulting in secondary neurodegenerative changes. By repairing the BSCB and/or removing the BSCB-derived injurious stimuli, we now identify that accumulation of blood-derived neurotoxic hemoglobin and iron in the spinal cord leads to early motor-neuron degeneration in SOD1(G93A) mice at least in part through iron-dependent oxidant stress. Using spontaneous or warfarin-accelerated microvascular lesions, motor-neuron dysfunction and injury were found to be proportional to the degree of BSCB disruption at early disease stages in SOD1(G93A) mice. Early treatment with an activated protein C analog restored BSCB integrity that developed from spontaneous or warfarin-accelerated microvascular lesions in SOD1(G93A) mice and eliminated neurotoxic hemoglobin and iron deposits. Restoration of BSCB integrity delayed onset of motor-neuron impairment and degeneration. Early chelation of blood-derived iron and antioxidant treatment mitigated early motor-neuronal injury. Our data suggest that BSCB breakdown contributes to early motor-neuron degeneration in ALS mice and that restoring BSCB integrity during an early disease phase retards the disease process.
Keywords: amyotrophic lateral sclerosis; neurodegeneration.
Conflict of interest statement
Conflict of interest statement: B.V.Z. is the scientific founder of ZZ Biotech, a start-up biotechnology company that is developing an activated protein C analog for stroke with possible implications in other neurological disorders. J.H.G. is a member of the Scientific Advisory Board of ZZ Biotech.
Figures



References
-
- Kiernan MC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. - PubMed
-
- Henkel JS, Beers DR, Wen S, Bowser R, Appel SH. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology. 2009;72(18):1614–1616. - PubMed
-
- Miyazaki K, et al. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosci Res. 2011;89(5):718–728. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 NS034467/NS/NINDS NIH HHS/United States
- R37 HL052246/HL/NHLBI NIH HHS/United States
- NS27036/NS/NINDS NIH HHS/United States
- NS34467/NS/NINDS NIH HHS/United States
- AG039452/AG/NIA NIH HHS/United States
- R01 HL052246/HL/NHLBI NIH HHS/United States
- AG23084/AG/NIA NIH HHS/United States
- R01 HL021544/HL/NHLBI NIH HHS/United States
- P01 HL031950/HL/NHLBI NIH HHS/United States
- HL052246/HL/NHLBI NIH HHS/United States
- 089701/Wellcome Trust/United Kingdom
- R01 AG023084/AG/NIA NIH HHS/United States
- R37 NS027036/NS/NINDS NIH HHS/United States
- P50 AG005142/AG/NIA NIH HHS/United States
- R01 AG039452/AG/NIA NIH HHS/United States
- R37 AG023084/AG/NIA NIH HHS/United States
- HL031950/HL/NHLBI NIH HHS/United States
- R01 NS027036/NS/NINDS NIH HHS/United States
- R01 NS034467/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
