

Genomic architecture of sickle cell disease in West African children
- PMID: 24592274
- PMCID: PMC3924578
- DOI: 10.3389/fgene.2014.00026
Genomic architecture of sickle cell disease in West African children
Abstract
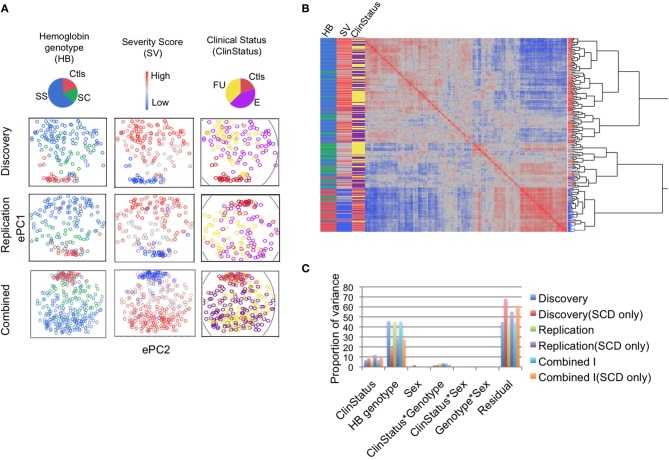
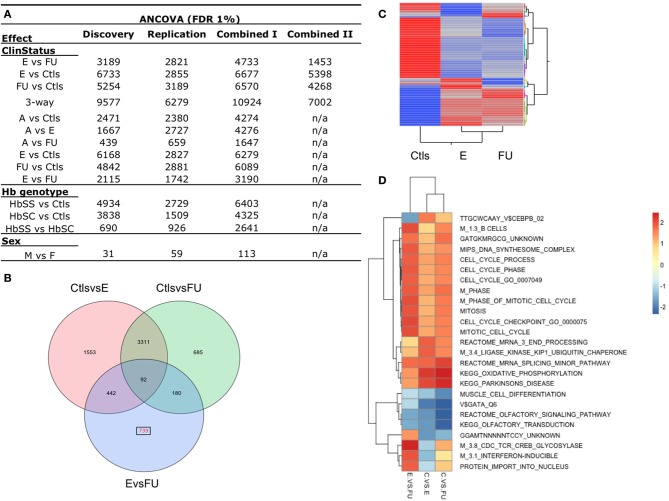
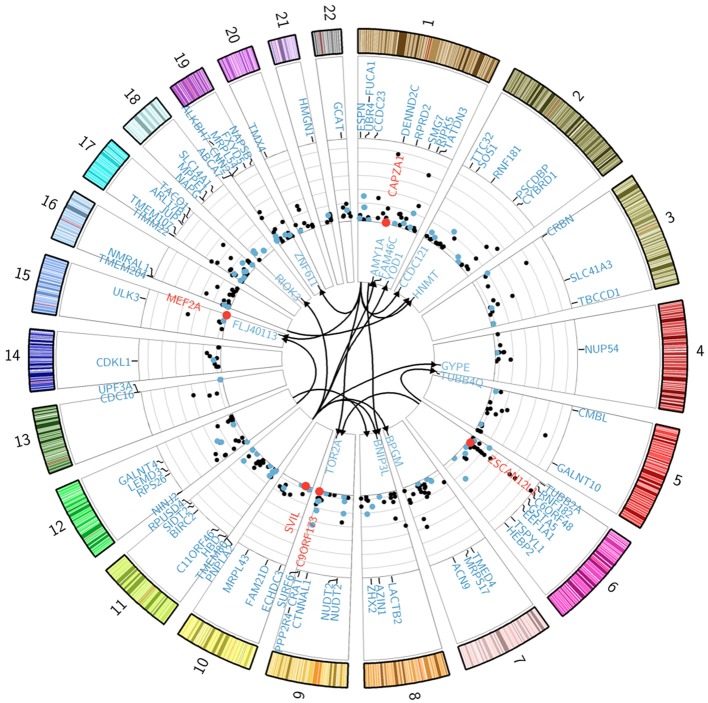
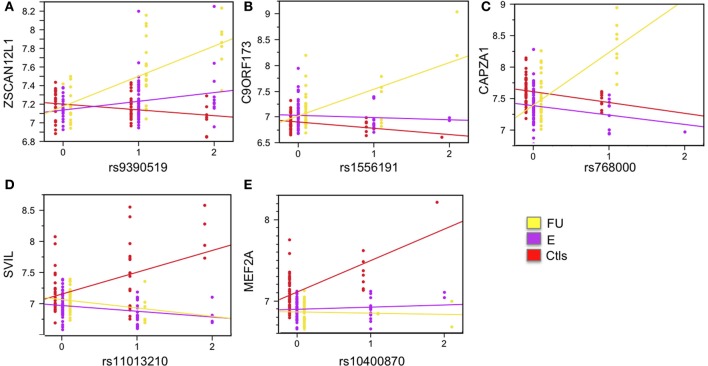
Sickle cell disease (SCD) is a congenital blood disease, affecting predominantly children from sub-Saharan Africa, but also populations world-wide. Although the causal mutation of SCD is known, the sources of clinical variability of SCD remain poorly understood, with only a few highly heritable traits associated with SCD having been identified. Phenotypic heterogeneity in the clinical expression of SCD is problematic for follow-up (FU), management, and treatment of patients. Here we used the joint analysis of gene expression and whole genome genotyping data to identify the genetic regulatory effects contributing to gene expression variation among groups of patients exhibiting clinical variability, as well as unaffected siblings, in Benin, West Africa. We characterized and replicated patterns of whole blood gene expression variation within and between SCD patients at entry to clinic, as well as in follow-up programs. We present a global map of genes involved in the disease through analysis of whole blood sampled from the cohort. Genome-wide association mapping of gene expression revealed 390 peak genome-wide significant expression SNPs (eSNPs) and 6 significant eSNP-by-clinical status interaction effects. The strong modulation of the transcriptome implicates pathways affecting core circulating cell functions and shows how genotypic regulatory variation likely contributes to the clinical variation observed in SCD.
Keywords: eSNP mapping; gene-by-environment interactions; genomics; sickle cell disease; transcriptome.
Figures
References
-
- Adams G. T., Snieder H., Mckie V. C., Clair B., Brambilla D., Adams R. J., et al. (2003). Genetic risk factors for cerebrovascular disease in children with sickle cell disease: design of a case-control association study and genomewide screen. BMC Med. Genet. 4:6 10.1186/1471-2350-4-6 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases