

ICAM-2 regulates vascular permeability and N-cadherin localization through ezrin-radixin-moesin (ERM) proteins and Rac-1 signalling
- PMID: 24593809
- PMCID: PMC4015342
- DOI: 10.1186/1478-811X-12-12
ICAM-2 regulates vascular permeability and N-cadherin localization through ezrin-radixin-moesin (ERM) proteins and Rac-1 signalling
Abstract
Background: Endothelial junctions control functions such as permeability, angiogenesis and contact inhibition. VE-Cadherin (VECad) is essential for the maintenance of intercellular contacts. In confluent endothelial monolayers, N-Cadherin (NCad) is mostly expressed on the apical and basal membrane, but in the absence of VECad it localizes at junctions. Both cadherins are required for vascular development. The intercellular adhesion molecule (ICAM)-2, also localized at endothelial junctions, is involved in leukocyte recruitment and angiogenesis.
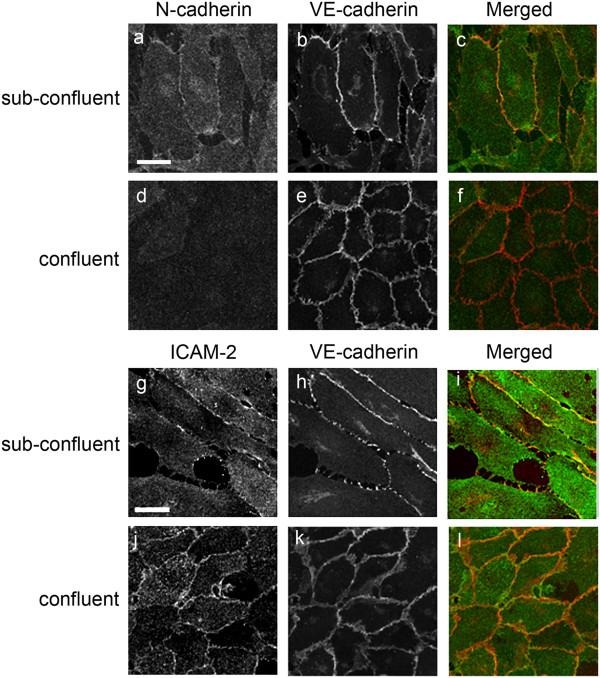
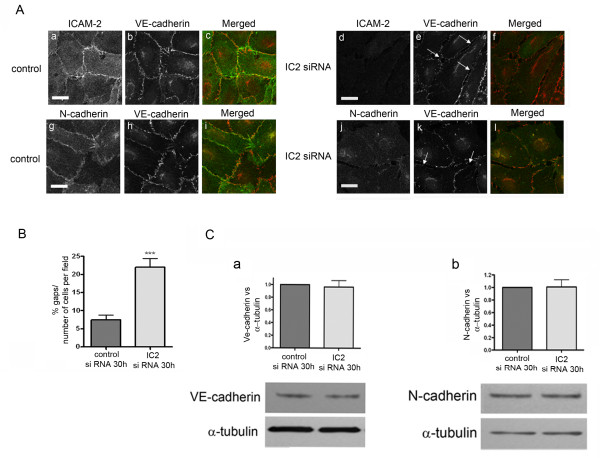
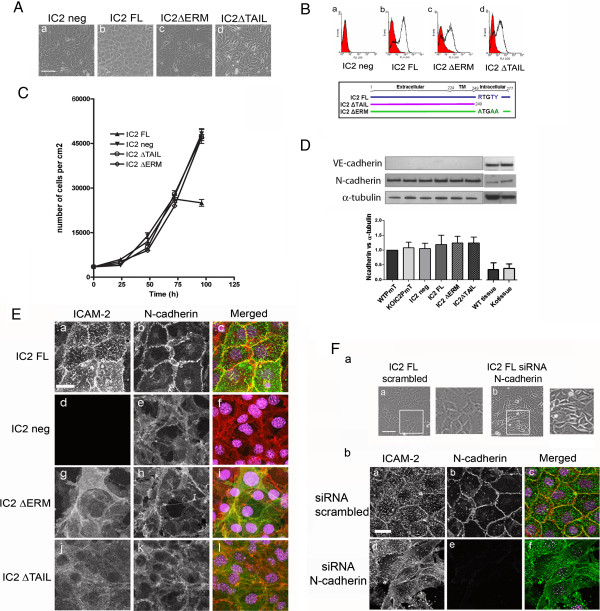
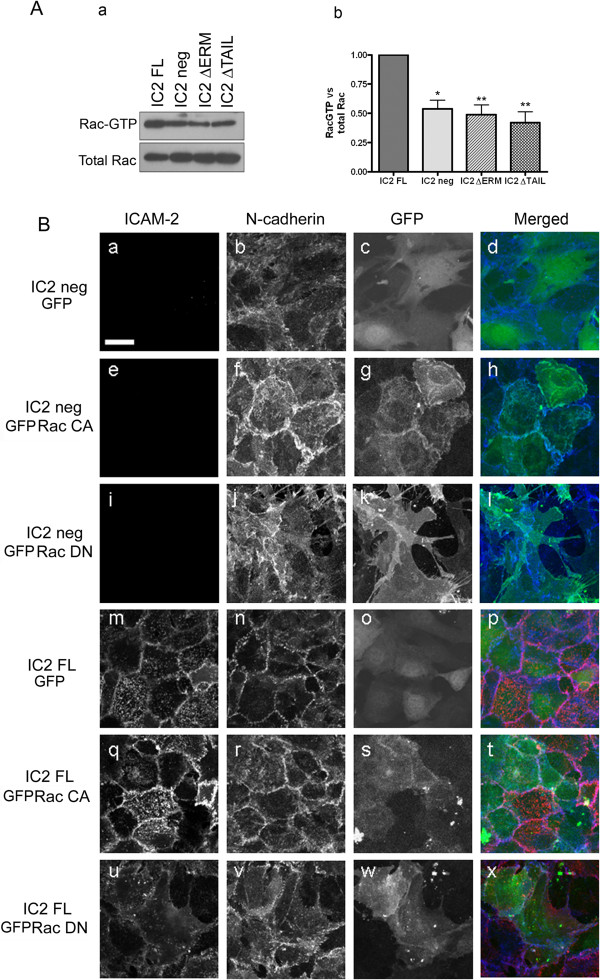
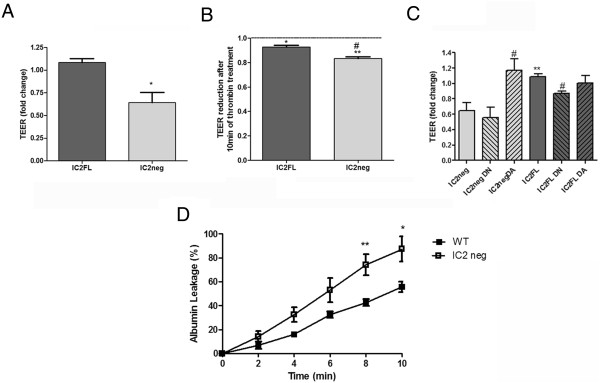
Results: In human umbilical vein endothelial cells (HUVEC), both VECad and NCad were found at nascent cell contacts of sub-confluent monolayers, but only VECad localized at the mature junctions of confluent monolayers. Inhibition of ICAM-2 expression by siRNA caused the appearance of small gaps at the junctions and a decrease in NCad junctional staining in sub-confluent monolayers. Endothelioma lines derived from WT or ICAM-2-deficient mice (IC2neg) lacked VECad and failed to form junctions, with loss of contact inhibition. Re-expression of full-length ICAM-2 (IC2 FL) in IC2neg cells restored contact inhibition through recruitment of NCad at the junctions. Mutant ICAM-2 lacking the binding site for ERM proteins (IC2 ΔERM) or the cytoplasmic tail (IC2 ΔTAIL) failed to restore junctions. ICAM-2-dependent Rac-1 activation was also decreased in these mutant cell lines. Barrier function, measured in vitro via transendothelial electrical resistance, was decreased in IC2neg cells, both in resting conditions and after thrombin stimulation. This was dependent on ICAM-2 signalling to the small GTPase Rac-1, since transendothelial electrical resistance of IC2neg cells was restored by constitutively active Rac-1. In vivo, thrombin-induced extravasation of FITC-labeled albumin measured by intravital fluorescence microscopy in the mouse cremaster muscle showed that permeability was increased in ICAM-2-deficient mice compared to controls.
Conclusions: These results indicate that ICAM-2 regulates endothelial barrier function and permeability through a pathway involving N-Cadherin, ERMs and Rac-1.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
