

Cell cycle, oncogenic and tumor suppressor pathways regulate numerous long and macro non-protein-coding RNAs
- PMID: 24594072
- PMCID: PMC4054595
- DOI: 10.1186/gb-2014-15-3-r48
Cell cycle, oncogenic and tumor suppressor pathways regulate numerous long and macro non-protein-coding RNAs
Abstract
Background: The genome is pervasively transcribed but most transcripts do not code for proteins, constituting non-protein-coding RNAs. Despite increasing numbers of functional reports of individual long non-coding RNAs (lncRNAs), assessing the extent of functionality among the non-coding transcriptional output of mammalian cells remains intricate. In the protein-coding world, transcripts differentially expressed in the context of processes essential for the survival of multicellular organisms have been instrumental in the discovery of functionally relevant proteins and their deregulation is frequently associated with diseases. We therefore systematically identified lncRNAs expressed differentially in response to oncologically relevant processes and cell-cycle, p53 and STAT3 pathways, using tiling arrays.
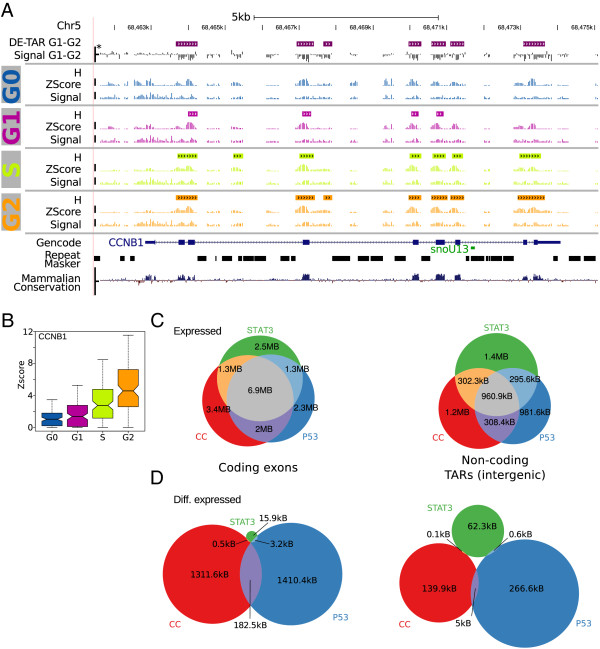
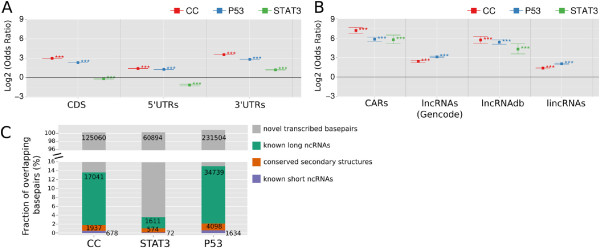
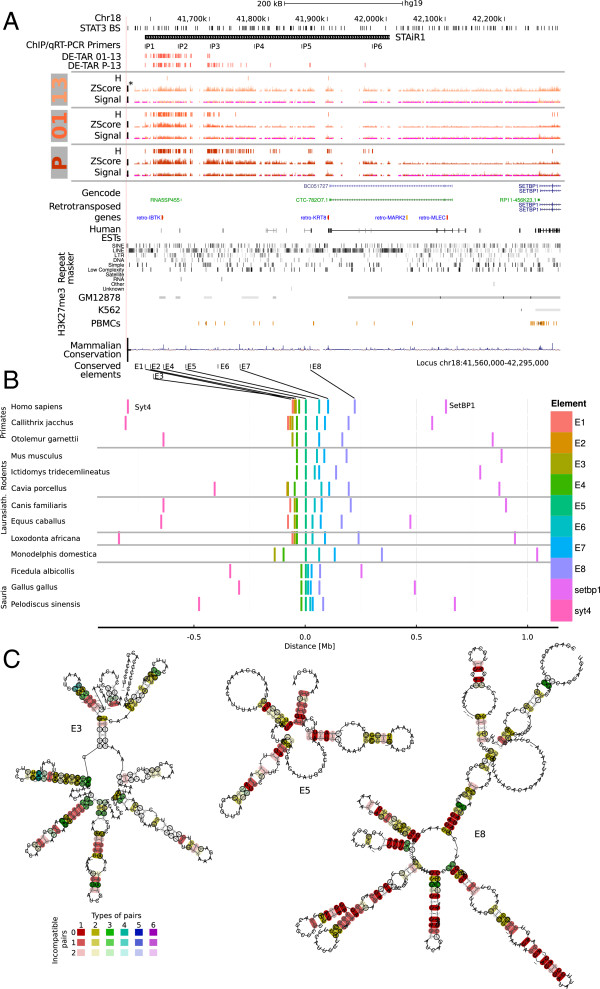
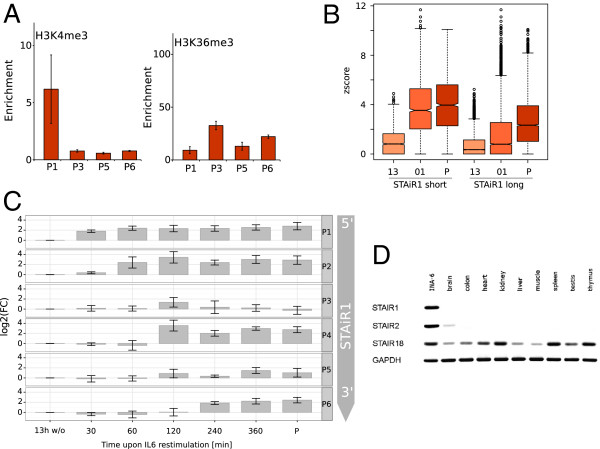
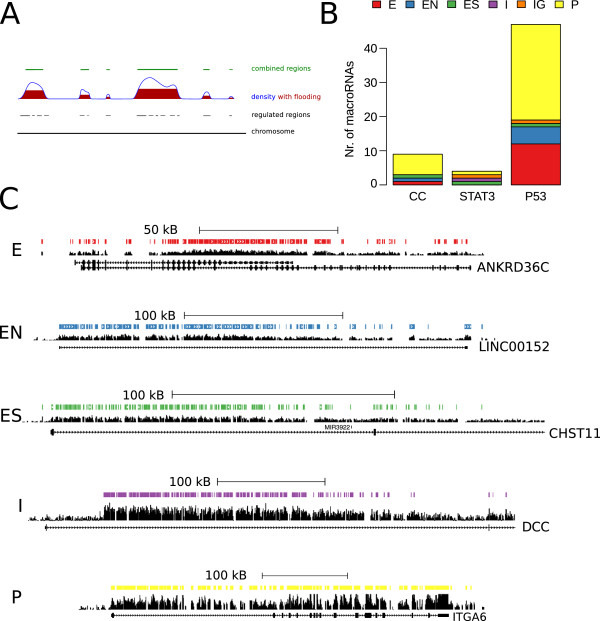
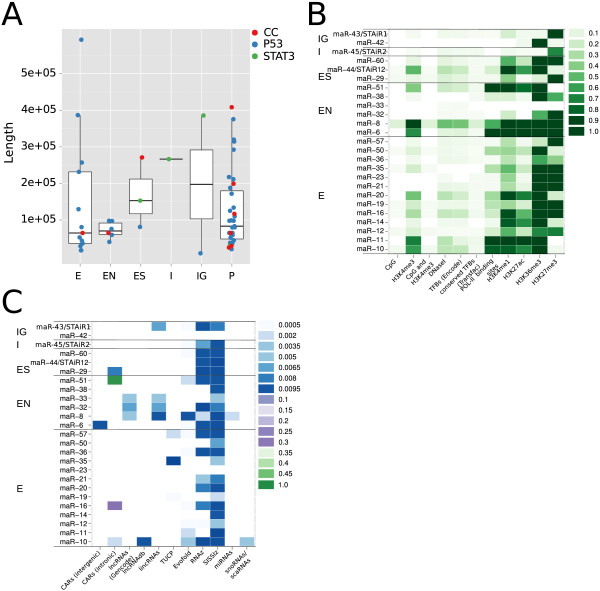
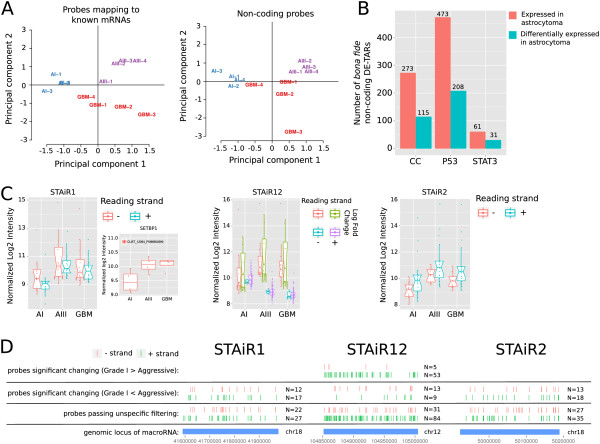
Results: We found that up to 80% of the pathway-triggered transcriptional responses are non-coding. Among these we identified very large macroRNAs with pathway-specific expression patterns and demonstrated that these are likely continuous transcripts. MacroRNAs contain elements conserved in mammals and sauropsids, which in part exhibit conserved RNA secondary structure. Comparing evolutionary rates of a macroRNA to adjacent protein-coding genes suggests a local action of the transcript. Finally, in different grades of astrocytoma, a tumor disease unrelated to the initially used cell lines, macroRNAs are differentially expressed.
Conclusions: It has been shown previously that the majority of expressed non-ribosomal transcripts are non-coding. We now conclude that differential expression triggered by signaling pathways gives rise to a similar abundance of non-coding content. It is thus unlikely that the prevalence of non-coding transcripts in the cell is a trivial consequence of leaky or random transcription events.
Figures
References
-
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest ARR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T. et al.The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. - PubMed
-
- Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15 Spec No 1:R17–R29. - PubMed
-
- Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8:413–423. - PubMed
-
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [ http://dx.doi.org/10.1038/nature11247] - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
