

A proteomic chronology of gene expression through the cell cycle in human myeloid leukemia cells
- PMID: 24596151
- PMCID: PMC3936288
- DOI: 10.7554/eLife.01630
A proteomic chronology of gene expression through the cell cycle in human myeloid leukemia cells
Abstract
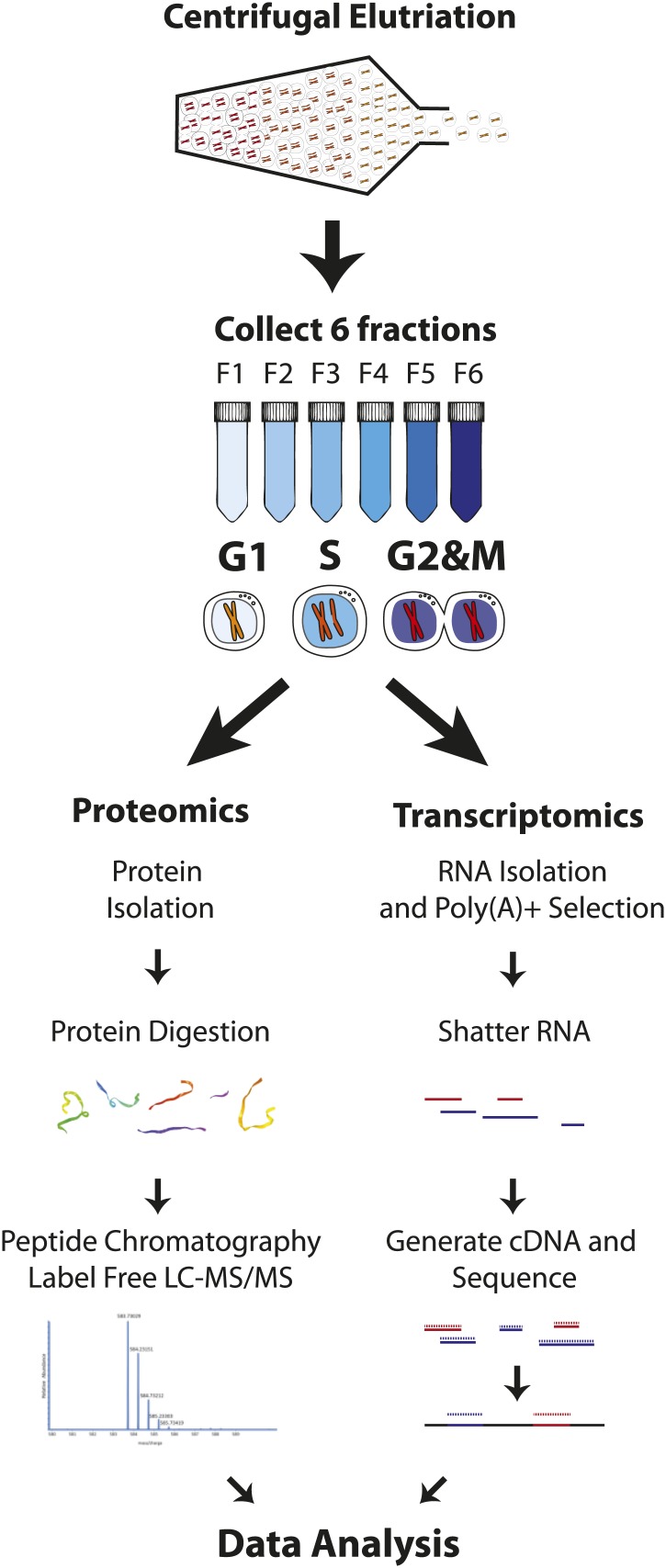
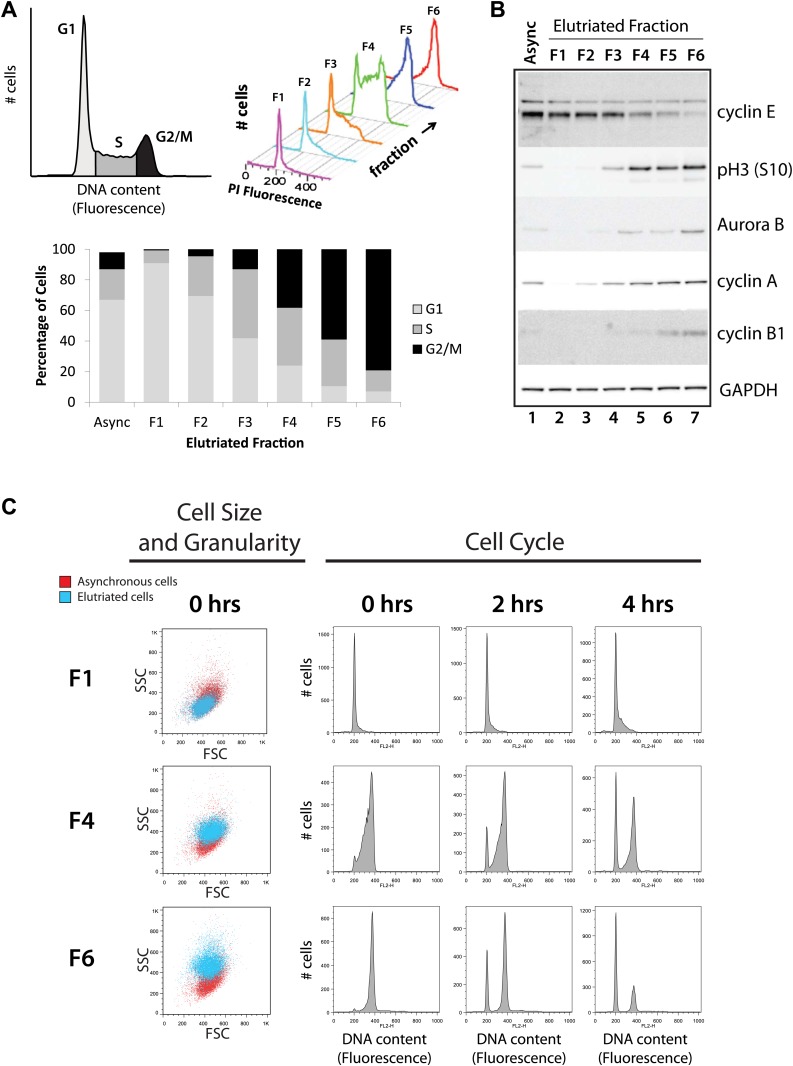
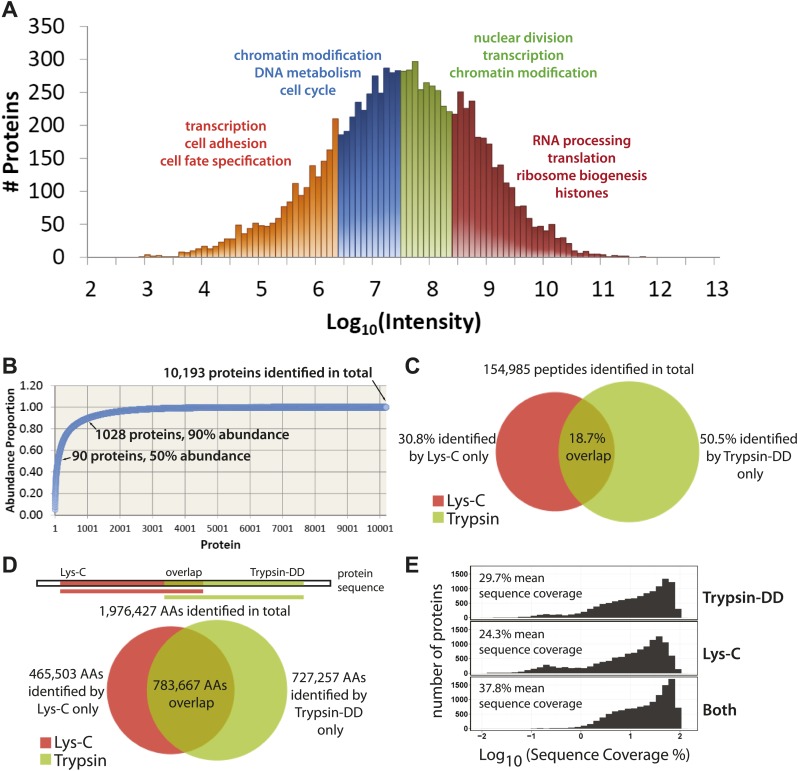
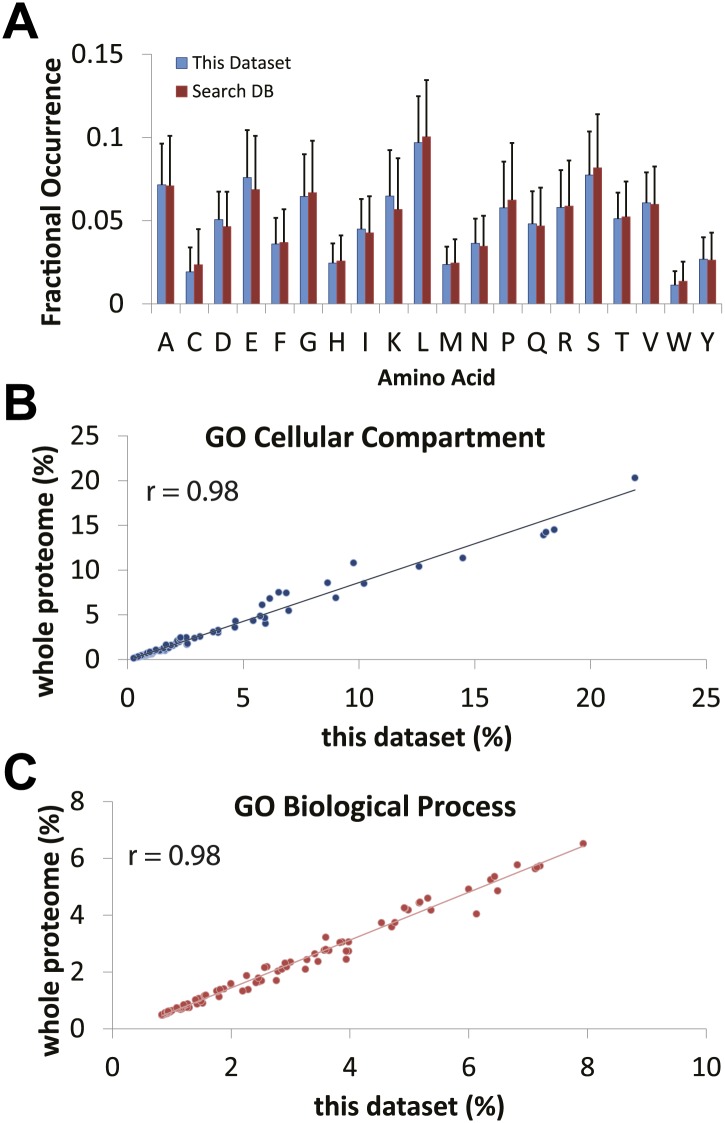
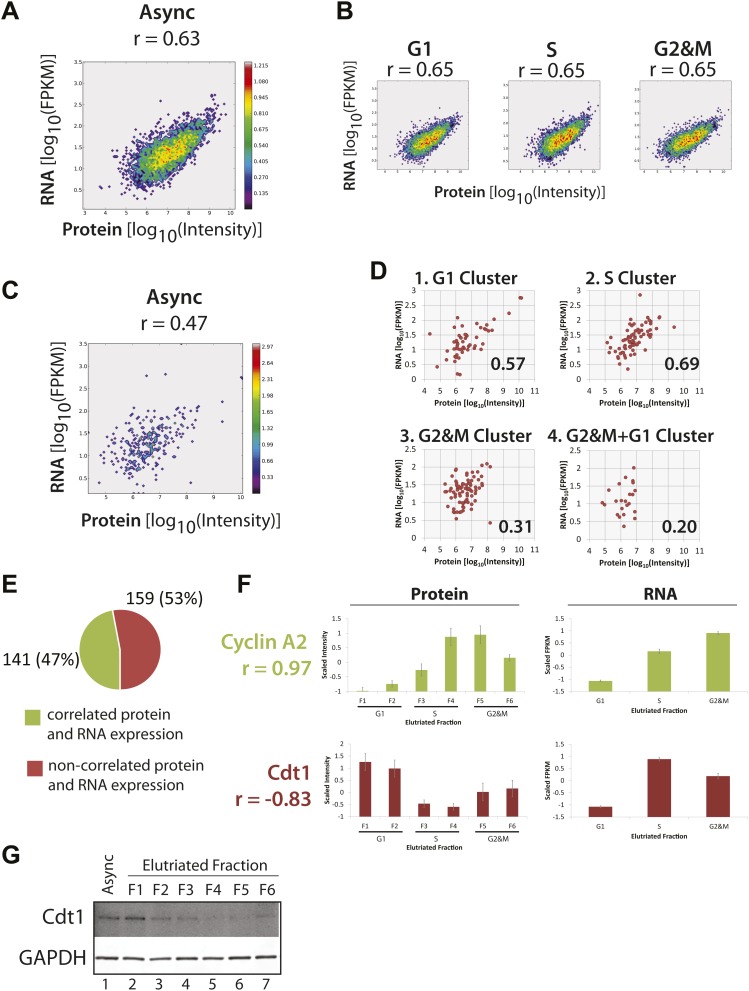
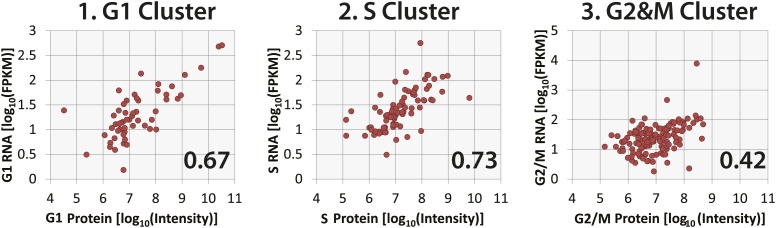
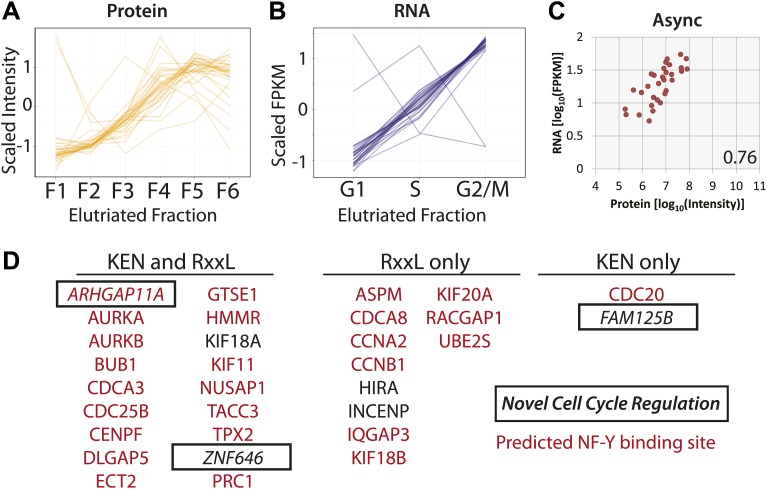
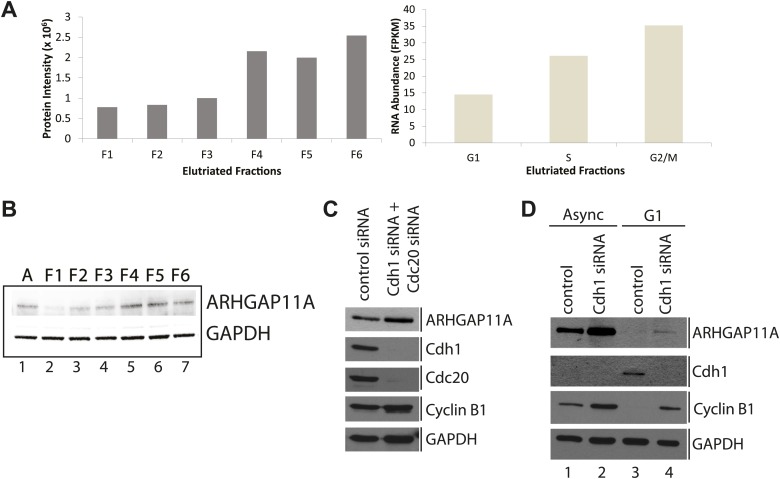
Technological advances have enabled the analysis of cellular protein and RNA levels with unprecedented depth and sensitivity, allowing for an unbiased re-evaluation of gene regulation during fundamental biological processes. Here, we have chronicled the dynamics of protein and mRNA expression levels across a minimally perturbed cell cycle in human myeloid leukemia cells using centrifugal elutriation combined with mass spectrometry-based proteomics and RNA-Seq, avoiding artificial synchronization procedures. We identify myeloid-specific gene expression and variations in protein abundance, isoform expression and phosphorylation at different cell cycle stages. We dissect the relationship between protein and mRNA levels for both bulk gene expression and for over ∼6000 genes individually across the cell cycle, revealing complex, gene-specific patterns. This data set, one of the deepest surveys to date of gene expression in human cells, is presented in an online, searchable database, the Encyclopedia of Proteome Dynamics (http://www.peptracker.com/epd/). DOI: http://dx.doi.org/10.7554/eLife.01630.001.
Keywords: RNA-Seq; cell cycle; mass spectrometry; proteomics; transcriptomics.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures


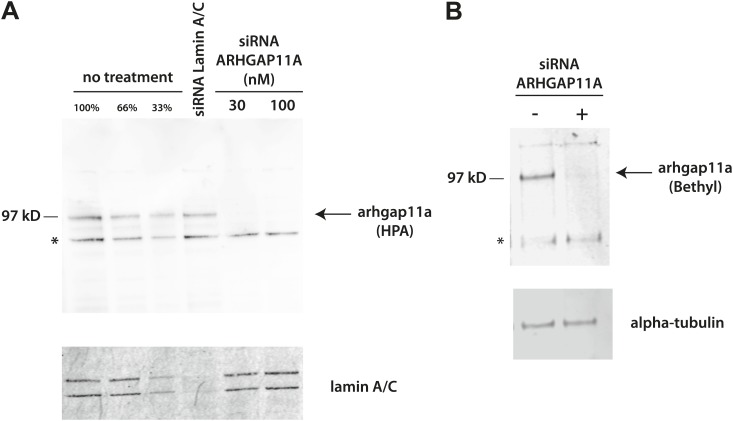
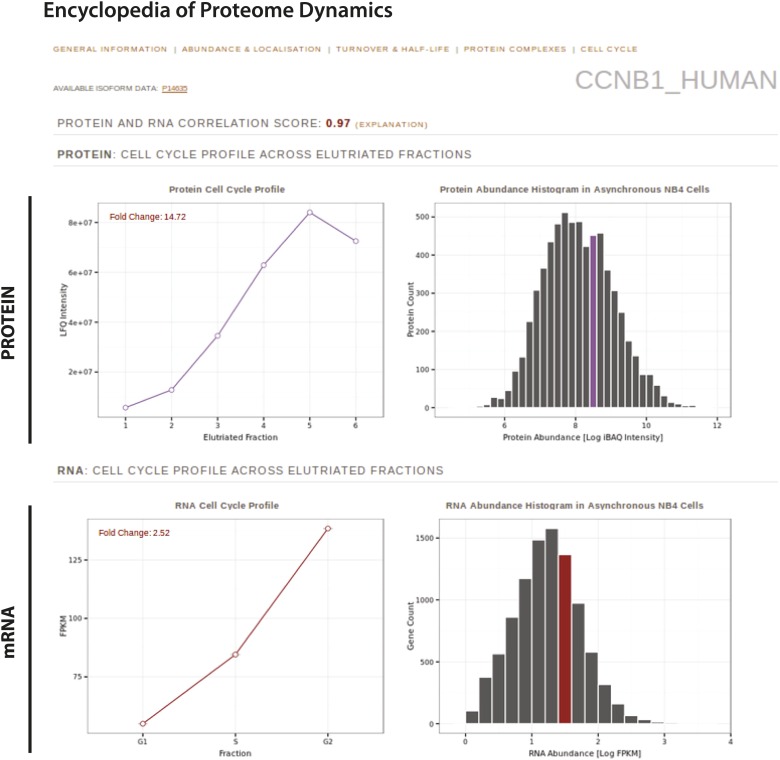
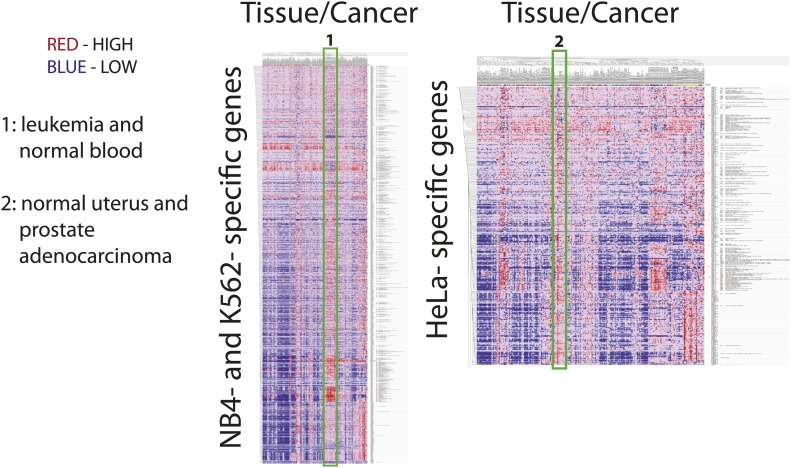
References
-
- Ballabeni A, Zamponi R, Moore JK, Helin K, Kirschner MW. 2013. Geminin deploys multiple mechanisms to regulate Cdt1 before cell division thus ensuring the proper execution of DNA replication. Proceedings of the National Academy of Sciences of the United States of America 110:E2848–E2853. 10.1073/pnas.1310677110 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
