

Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria
- PMID: 24598713
- PMCID: PMC3962989
- DOI: 10.1212/WNL.0000000000000241
Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria
Abstract
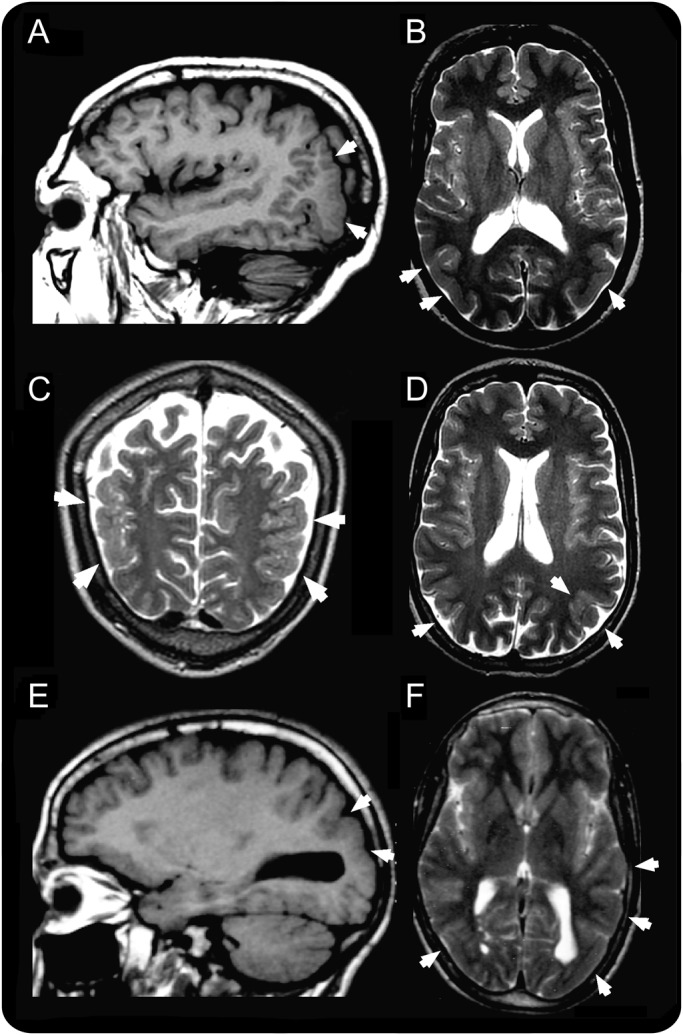
Objective: The aim of this study was to identify the causal gene in a consanguineous Moroccan family with temporo-occipital polymicrogyria, psychiatric manifestations, and epilepsy, previously mapped to the 6q16-q22 region.
Methods: We used exome sequencing and analyzed candidate variants in the 6q16-q22 locus, as well as a rescue assay in Fig4-null mouse fibroblasts and immunohistochemistry of Fig4-null mouse brains.
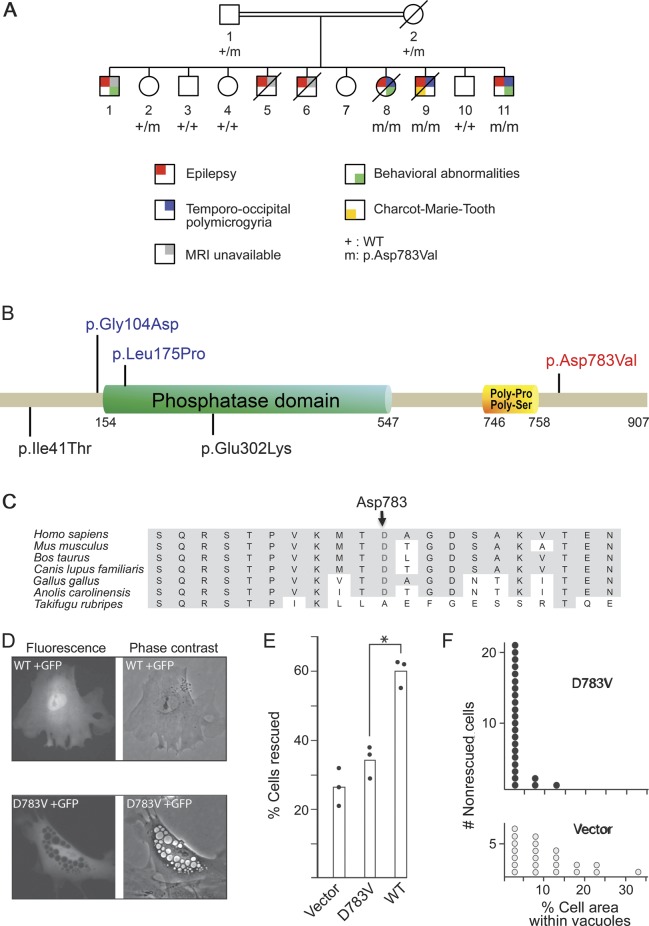
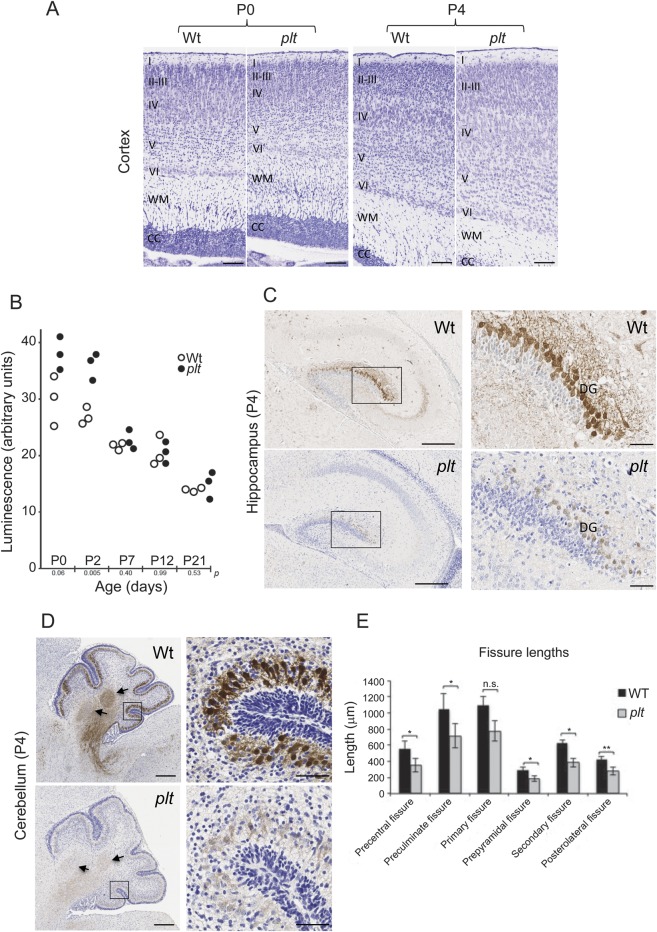
Results: A homozygous missense mutation (p.Asp783Val) in the phosphoinositide phosphatase gene FIG4 was identified. Pathogenicity of the variant was supported by impaired rescue of the enlarged vacuoles in transfected fibroblasts from Fig4-deficient mice. Histologic examination of Fig4-null mouse brain revealed neurodevelopmental impairment in the hippocampus, cortex, and cerebellum as well as impaired cerebellar gyration/foliation reminiscent of human cortical malformations.
Conclusions: This study extends the spectrum of phenotypes associated with FIG4 mutations to include cortical malformation associated with seizures and psychiatric manifestations, in addition to the previously described Charcot-Marie-Tooth disease type 4J and Yunis-Varón syndrome.
Figures
References
-
- Piao X, Hill RS, Bodell A, et al. G protein-coupled receptor-dependent development of human frontal cortex. Science 2004;303:2033–2036 - PubMed
-
- Valence S, Poirier K, Lebrun N, et al. Homozygous truncating mutation of the KBP gene, encoding a KIF1B-binding protein, in a familial case of fetal polymicrogyria. Neurogenetics 2013;14:215–224 - PubMed
-
- Borck G, Wunram H, Steiert A, et al. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum Genet 2011;129:45–50 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases