

Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1
- PMID: 24599401
- PMCID: PMC4065158
- DOI: 10.1093/hmg/ddu101
Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1
Abstract
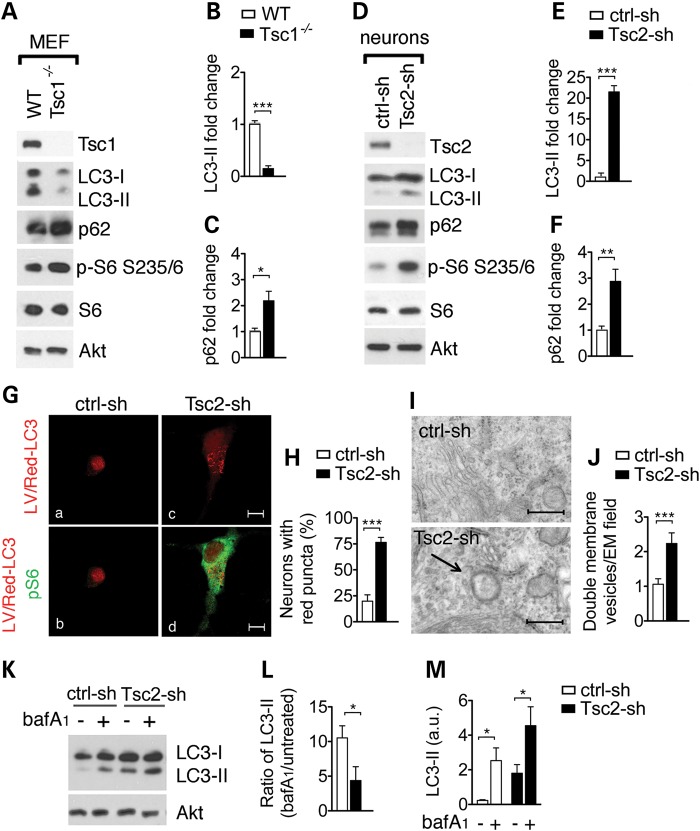
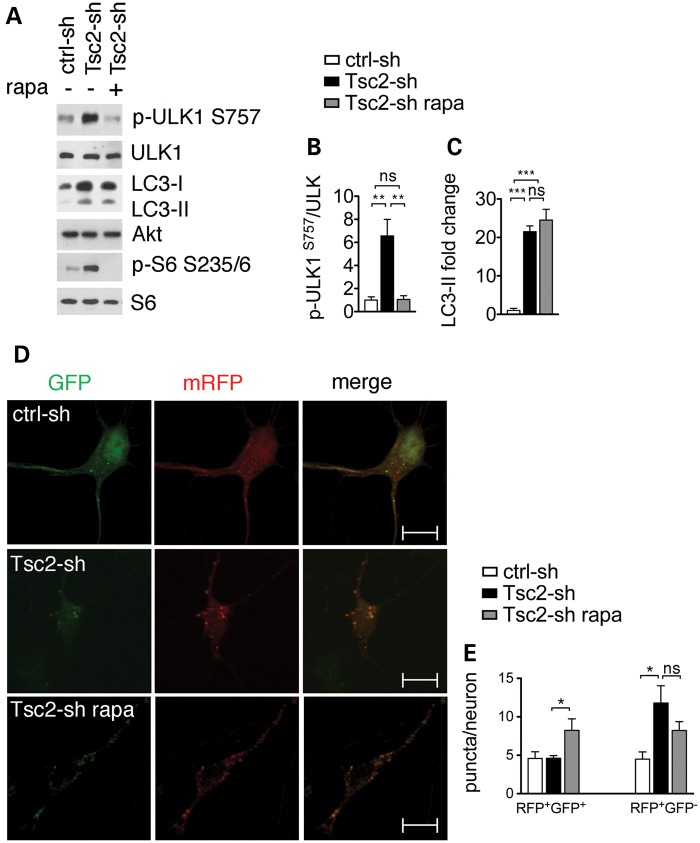
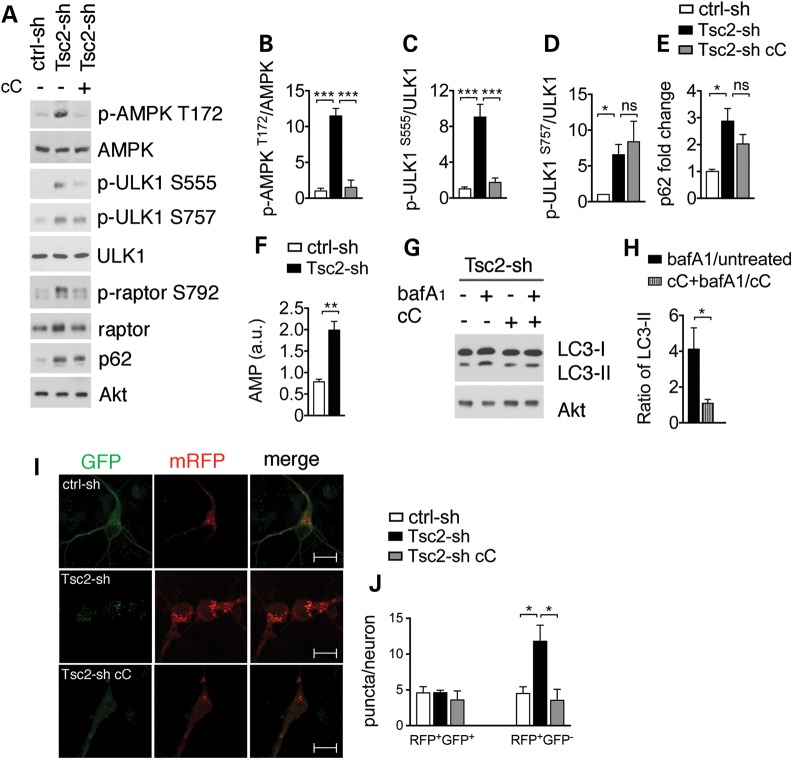
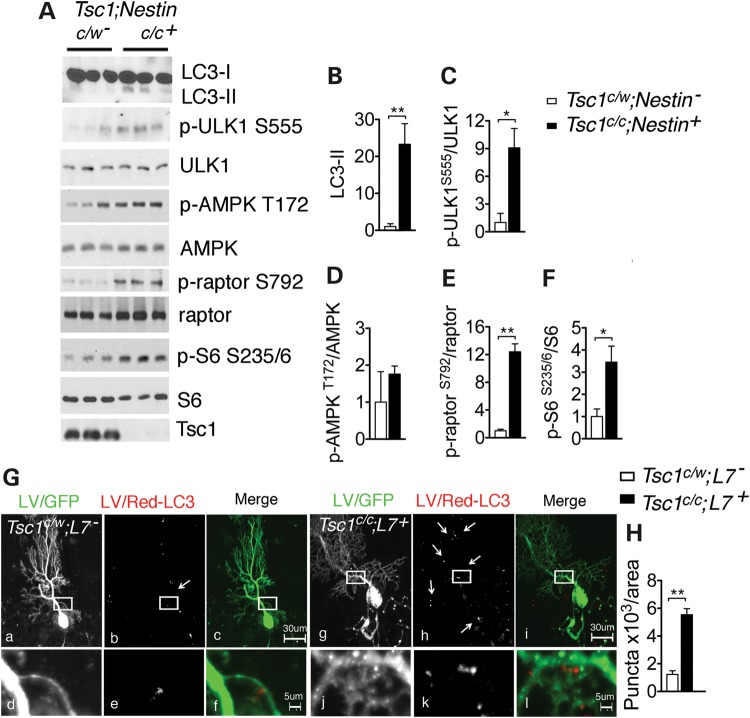
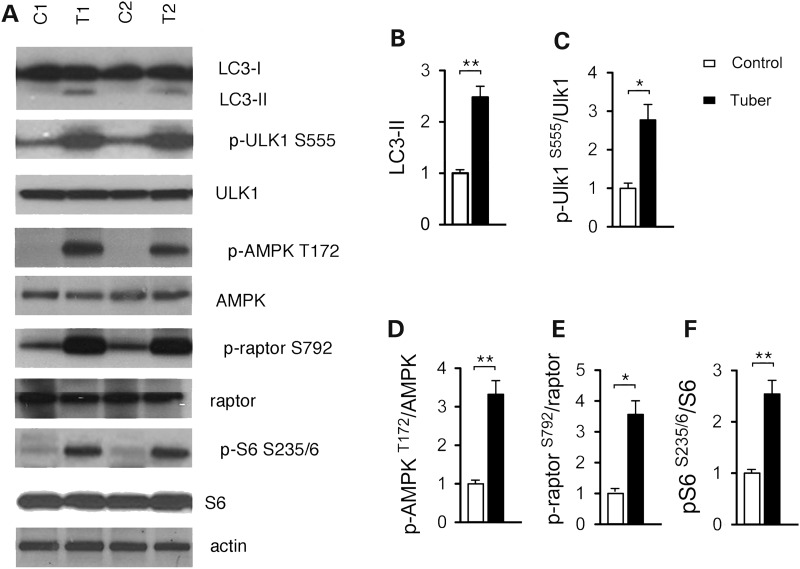
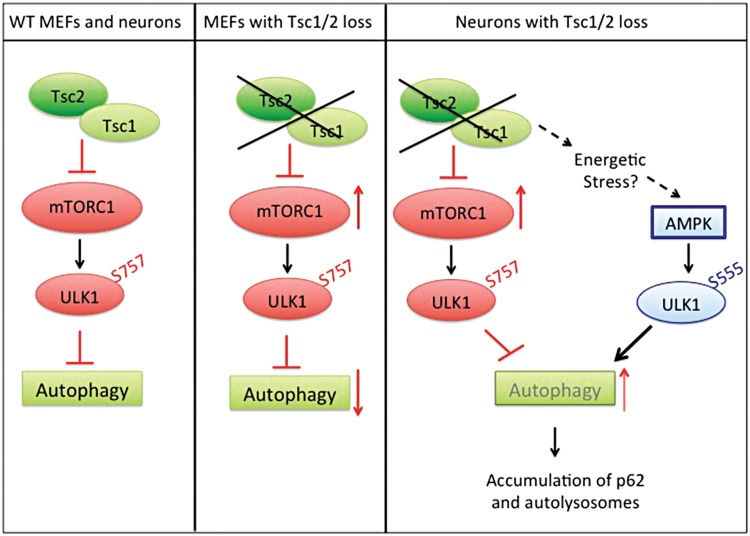
Tuberous sclerosis complex (TSC) is a disorder arising from mutation in the TSC1 or TSC2 gene, characterized by the development of hamartomas in various organs and neurological manifestations including epilepsy, intellectual disability and autism. TSC1/2 protein complex negatively regulates the mammalian target of rapamycin complex 1 (mTORC1) a master regulator of protein synthesis, cell growth and autophagy. Autophagy is a cellular quality-control process that sequesters cytosolic material in double membrane vesicles called autophagosomes and degrades it in autolysosomes. Previous studies in dividing cells have shown that mTORC1 blocks autophagy through inhibition of Unc-51-like-kinase1/2 (ULK1/2). Despite the fact that autophagy plays critical roles in neuronal homeostasis, little is known on the regulation of autophagy in neurons. Here we show that unlike in non-neuronal cells, Tsc2-deficient neurons have increased autolysosome accumulation and autophagic flux despite mTORC1-dependent inhibition of ULK1. Our data demonstrate that loss of Tsc2 results in autophagic activity via AMPK-dependent activation of ULK1. Thus, in Tsc2-knockdown neurons AMPK activation is the dominant regulator of autophagy. Notably, increased AMPK activity and autophagy activation are also found in the brains of Tsc1-conditional mouse models and in cortical tubers resected from TSC patients. Together, our findings indicate that neuronal Tsc1/2 complex activity is required for the coordinated regulation of autophagy by AMPK. By uncovering the autophagy dysfunction associated with Tsc2 loss in neurons, our work sheds light on a previously uncharacterized cellular mechanism that contributes to altered neuronal homeostasis in TSC disease.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
