

Treatment strategies for inherited optic neuropathies: past, present and future
- PMID: 24603424
- PMCID: PMC4017118
- DOI: 10.1038/eye.2014.37
Treatment strategies for inherited optic neuropathies: past, present and future
Abstract
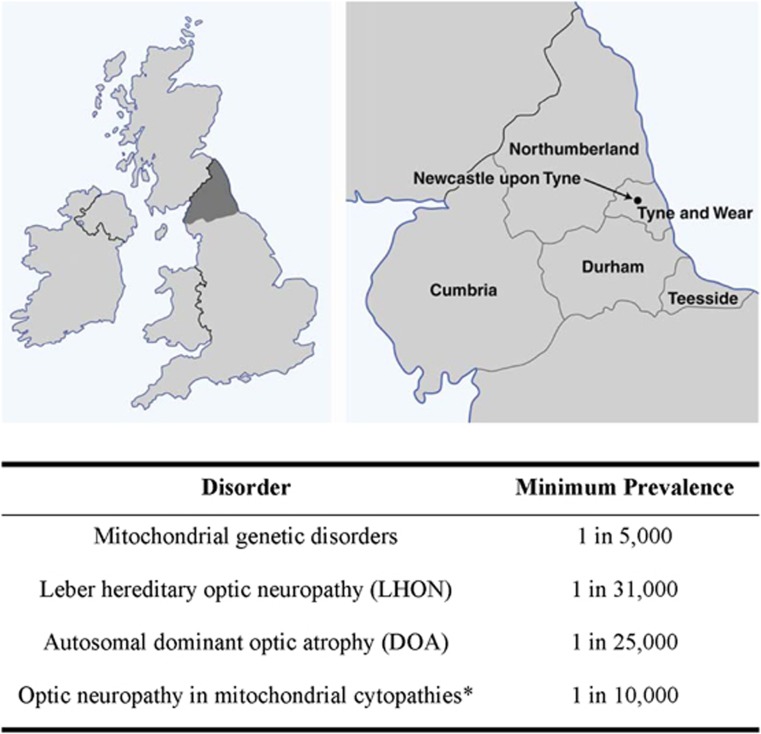
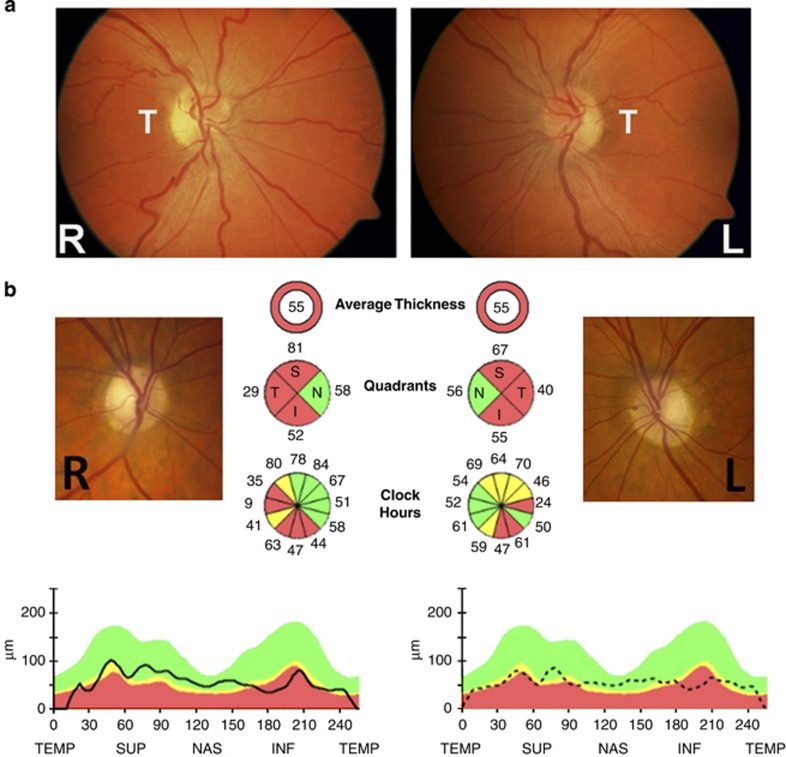
Bilateral visual loss secondary to inherited optic neuropathies is an important cause of registrable blindness among children and young adults. The two prototypal disorders seen in clinical practice are Leber hereditary optic neuropathy (LHON) and autosomal dominant optic atrophy (DOA). About 90% of LHON cases are due to one of three mitochondrial DNA (mtDNA) point mutations: m.3460G>A, m.11778G>A, and m.14484T>C, which affect critical complex I subunits of the mitochondrial respiratory chain. The majority of patients with DOA harbour pathogenic mutations within OPA1, a nuclear gene that codes for a multifunctional inner mitochondrial membrane protein. Despite their contrasting genetic basis, LHON and DOA share overlapping pathological and clinical features that serve to highlight the striking tissue-specific vulnerability of the retinal ganglion cell (RGC) layer to disturbed mitochondrial function. In addition to severe visual loss secondary to progressive optic nerve degeneration, a subgroup of patients will also develop a more aggressive syndromic phenotype marked by significant neurological deficits. The management of LHON and DOA remains largely supportive, but major advances in our understanding of the mechanisms underpinning RGC loss in these two disorders are paving the way for novel forms of treatment aimed at halting or reversing visual deterioration at different stages of the disease process. In addition to neuroprotective strategies for rescuing RGCs from irreversible cell death, innovative in vitro fertilisation techniques are providing the tantalising prospect of preventing the germline transmission of pathogenic mtDNA mutations, eradicating in so doing the risk of disease in future generations.
Figures
References
-
- Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retinal Eye Res. 2004;23 (1:53–89. - PubMed
-
- DiMauro S, Schon EA. Mechanisms of disease: Mitochondrial respiratory-chain diseases. New Engl J Med. 2003;348 (26:2656–2668. - PubMed
-
- Schapira AHV. Mitochondrial diseases. Lancet. 2012;379 (9828:1825–1834. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
