

Common genetic variants modulate pathogen-sensing responses in human dendritic cells
- PMID: 24604203
- PMCID: PMC4124741
- DOI: 10.1126/science.1246980
Common genetic variants modulate pathogen-sensing responses in human dendritic cells
Abstract
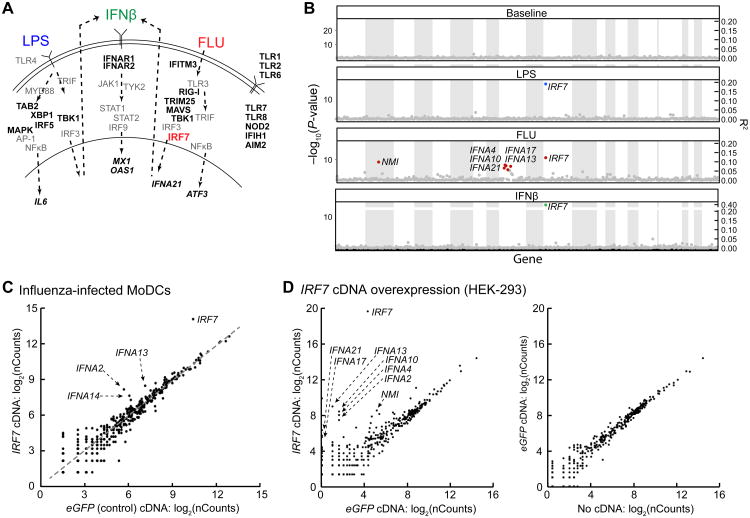
Little is known about how human genetic variation affects the responses to environmental stimuli in the context of complex diseases. Experimental and computational approaches were applied to determine the effects of genetic variation on the induction of pathogen-responsive genes in human dendritic cells. We identified 121 common genetic variants associated in cis with variation in expression responses to Escherichia coli lipopolysaccharide, influenza, or interferon-β (IFN-β). We localized and validated causal variants to binding sites of pathogen-activated STAT (signal transducer and activator of transcription) and IRF (IFN-regulatory factor) transcription factors. We also identified a common variant in IRF7 that is associated in trans with type I IFN induction in response to influenza infection. Our results reveal common alleles that explain interindividual variation in pathogen sensing and provide functional annotation for genetic variants that alter susceptibility to inflammatory diseases.
Figures





Comment in
-
Genetics. A genomic road map for complex human disease.Science. 2014 Mar 7;343(6175):1087-8. doi: 10.1126/science.1251426. Science. 2014. PMID: 24604188 No abstract available.
-
Immunogenetics: tracking immune activity across the genome.Nat Rev Immunol. 2014 Apr;14(4):212. doi: 10.1038/nri3653. Nat Rev Immunol. 2014. PMID: 24662379 No abstract available.
References
-
- Schadt EE, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003 Mar 20;422:297. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- R01 HG004037/HG/NHGRI NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- R01 AI091568/AI/NIAID NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- DP1 CA174427/CA/NCI NIH HHS/United States
- F32 AG043267/AG/NIA NIH HHS/United States
- U19 AI082630/AI/NIAID NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- DP2 OD002230/OD/NIH HHS/United States
- DP1 MH100706/MH/NIMH NIH HHS/United States
- P50 HG006193/HG/NHGRI NIH HHS/United States
- RC2 GM093080/GM/NIGMS NIH HHS/United States
- DP1 MH100706/DP/NCCDPHP CDC HHS/United States
- R01 AR063759/AR/NIAMS NIH HHS/United States
- R01 DK097768/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
