

General approach for in vivo recovery of cell type-specific effector gene sets
- PMID: 24604781
- PMCID: PMC4009615
- DOI: 10.1101/gr.167668.113
General approach for in vivo recovery of cell type-specific effector gene sets
Abstract
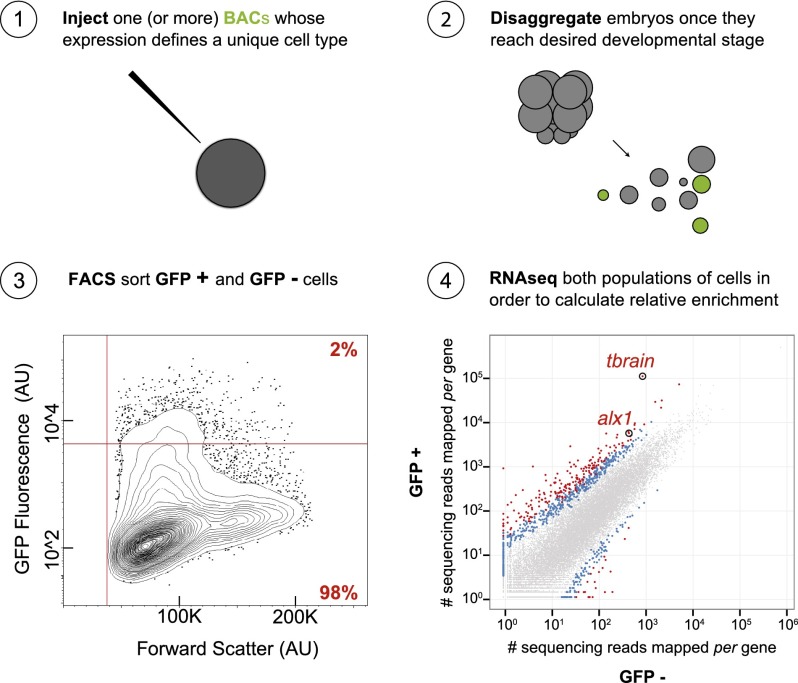
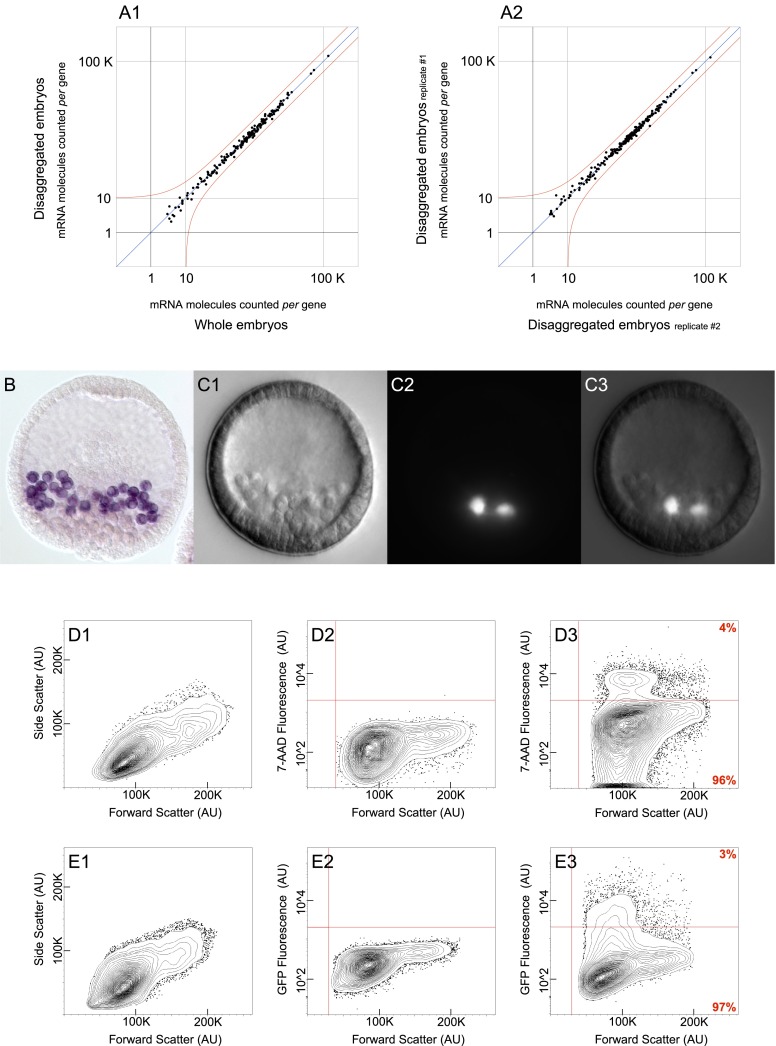
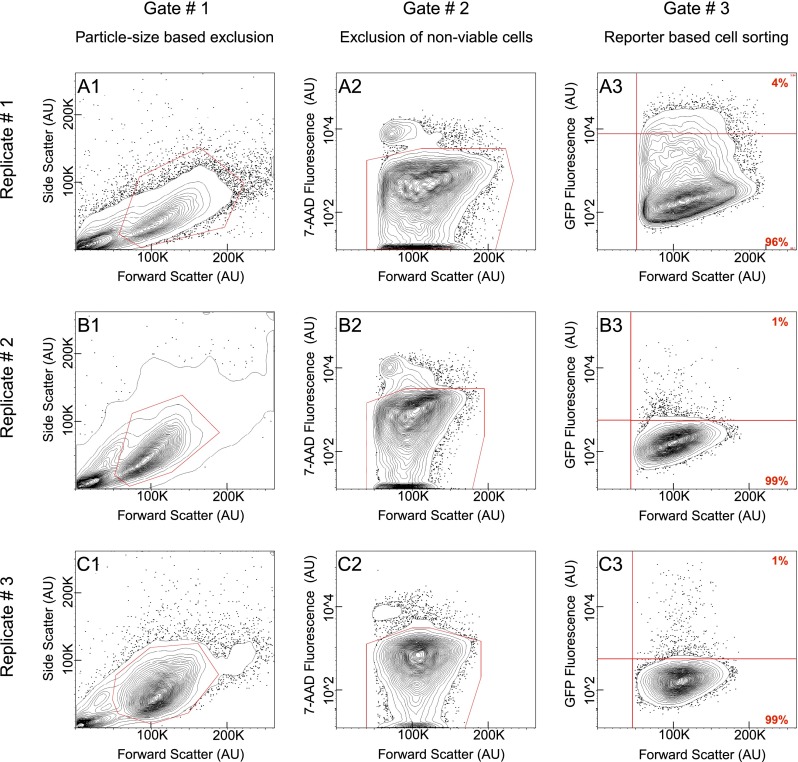
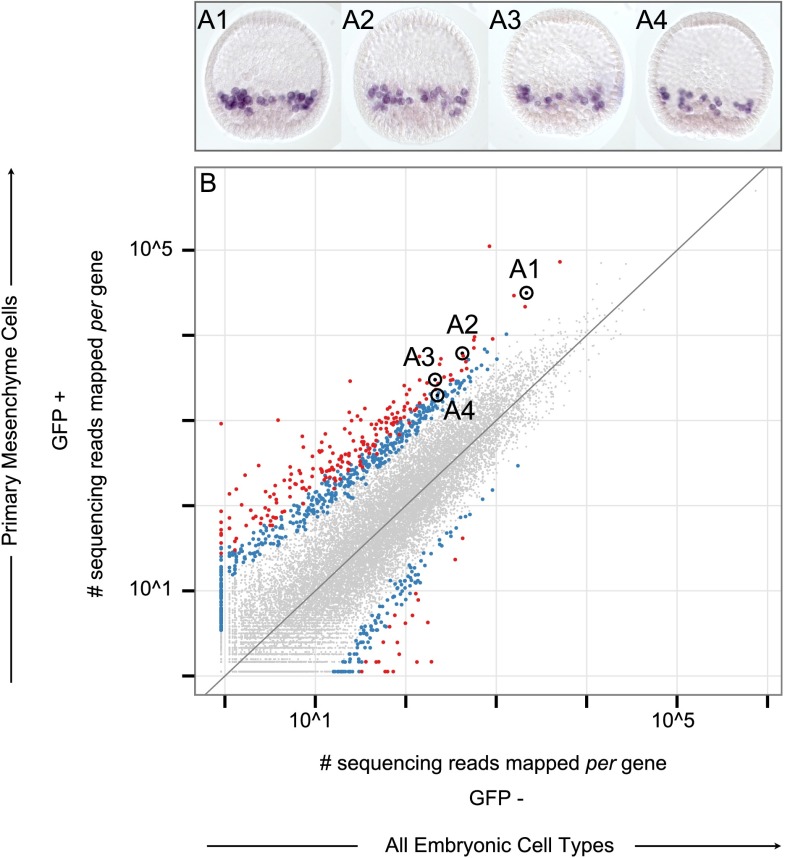
Differentially expressed, cell type-specific effector gene sets hold the key to multiple important problems in biology, from theoretical aspects of developmental gene regulatory networks (GRNs) to various practical applications. Although individual cell types of interest have been recovered by various methods and analyzed, systematic recovery of multiple cell type-specific gene sets from whole developing organisms has remained problematic. Here we describe a general methodology using the sea urchin embryo, a material of choice because of the large-scale GRNs already solved for this model system. This method utilizes the regulatory states expressed by given cells of the embryo to define cell type and includes a fluorescence activated cell sorting (FACS) procedure that results in no perturbation of transcript representation. We have extensively validated the method by spatial and qualitative analyses of the transcriptome expressed in isolated embryonic skeletogenic cells and as a consequence, generated a prototypical cell type-specific transcriptome database.
Figures

References
-
- Davidson EH. 2006. The regulatory genome. Academic, Burlington, MA.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous