

Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases
- PMID: 24606804
- PMCID: PMC4823001
- DOI: 10.2174/1381612820666140305224906
Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases
Abstract
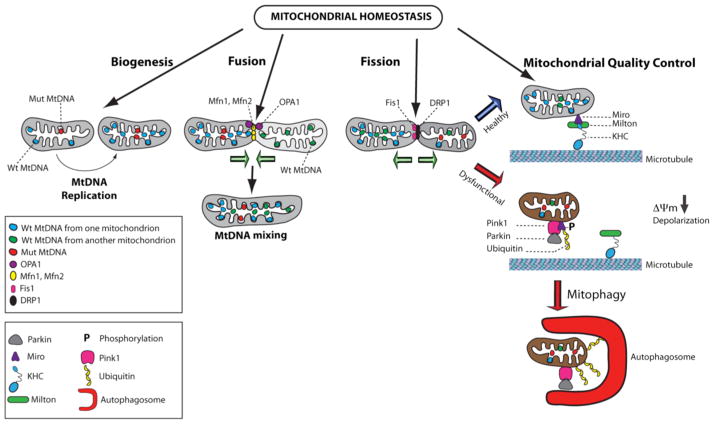
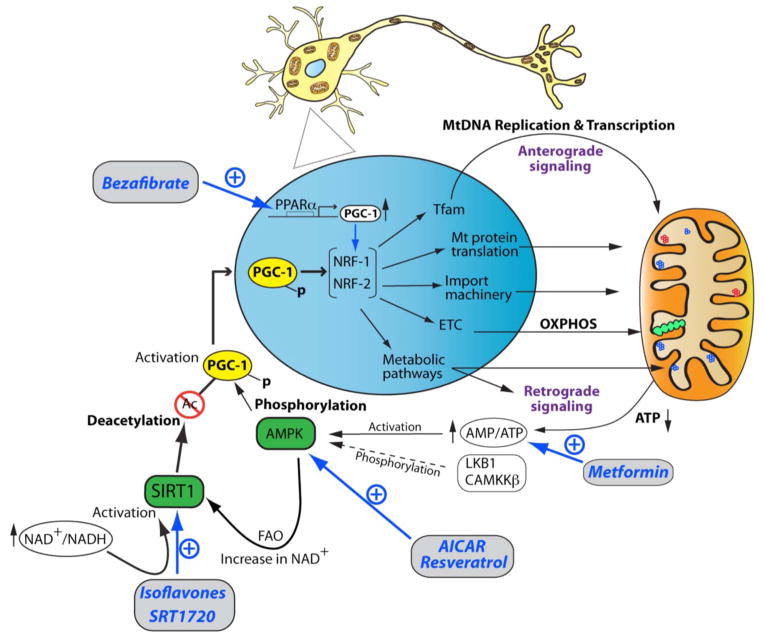
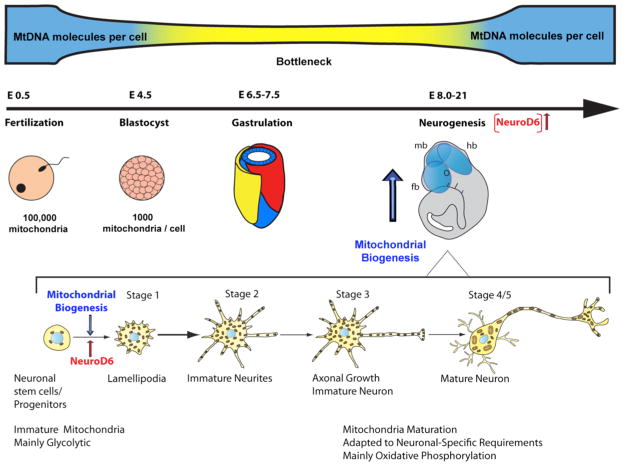
In the developing and mature brain, mitochondria act as central hubs for distinct but interwined pathways, necessary for neural development, survival, activity, connectivity and plasticity. In neurons, mitochondria assume diverse functions, such as energy production in the form of ATP, calcium buffering and generation of reactive oxygen species. Mitochondrial dysfunction contributes to a range of neurodevelopmental and neurodegenerative diseases, making mitochondria a potential target for pharmacological-based therapies. Pathogenesis associated with these diseases is accompanied by an increase in mitochondrial mass, a quantitative increase to overcome a qualitative deficiency due to mutated mitochondrial proteins that are either nuclear- or mitochondrial-encoded. This compensatory biological response is maladaptive, as it fails to sufficiently augment the bioenergetically functional mitochondrial mass and correct for the ATP deficit. Since regulation of neuronal mitochondrial biogenesis has been scantily investigated, our current understanding on the network of transcriptional regulators, co-activators and signaling regulators mainly derives from other cellular systems. The purpose of this review is to present the current state of our knowledge and understanding of the transcriptional and signaling cascades controlling neuronal mitochondrial biogenesis and the various therapeutic approaches to enhance the functional mitochondrial mass in the context of neurodevelopmental disorders and adult-onset neurodegenerative diseases.
Conflict of interest statement
The authors confirm that this article content has no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
