

Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness
- PMID: 24606897
- PMCID: PMC3969850
- DOI: 10.1016/j.cmet.2014.01.019
Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness
Abstract
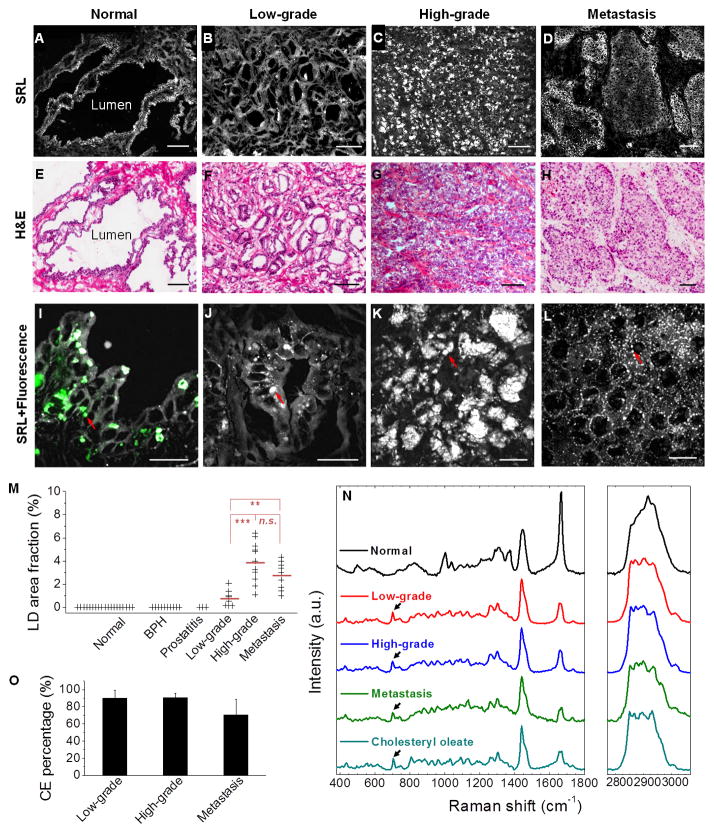
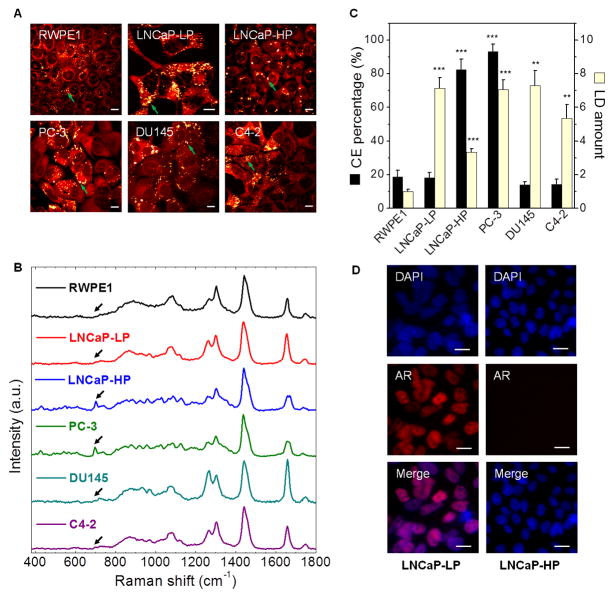
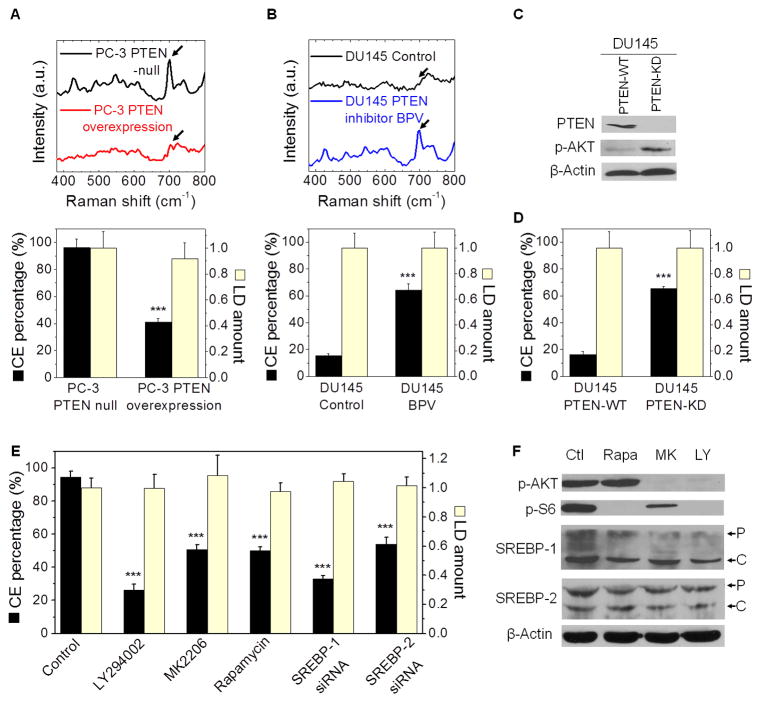
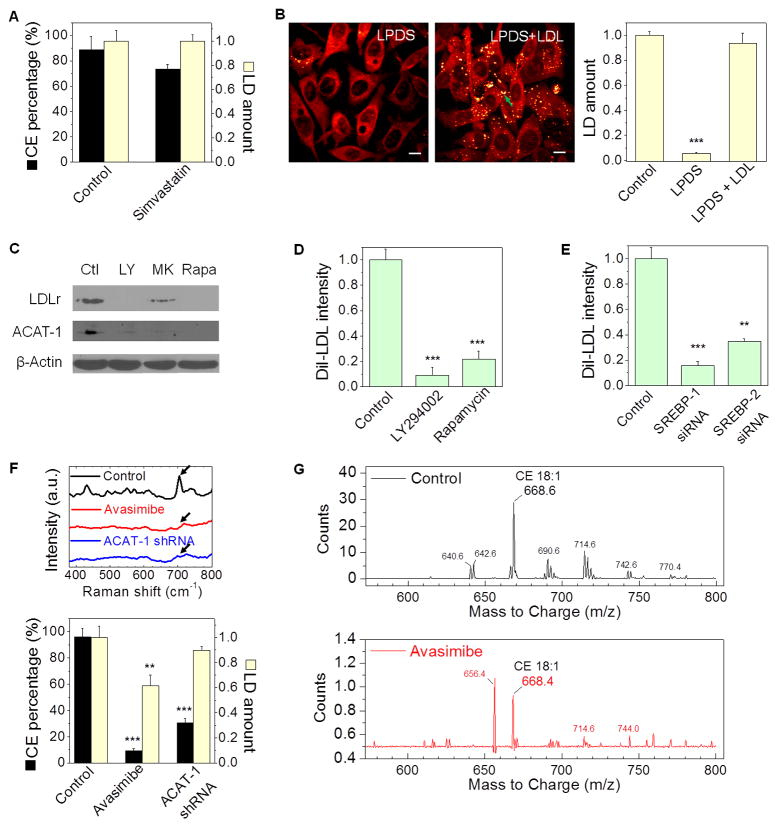
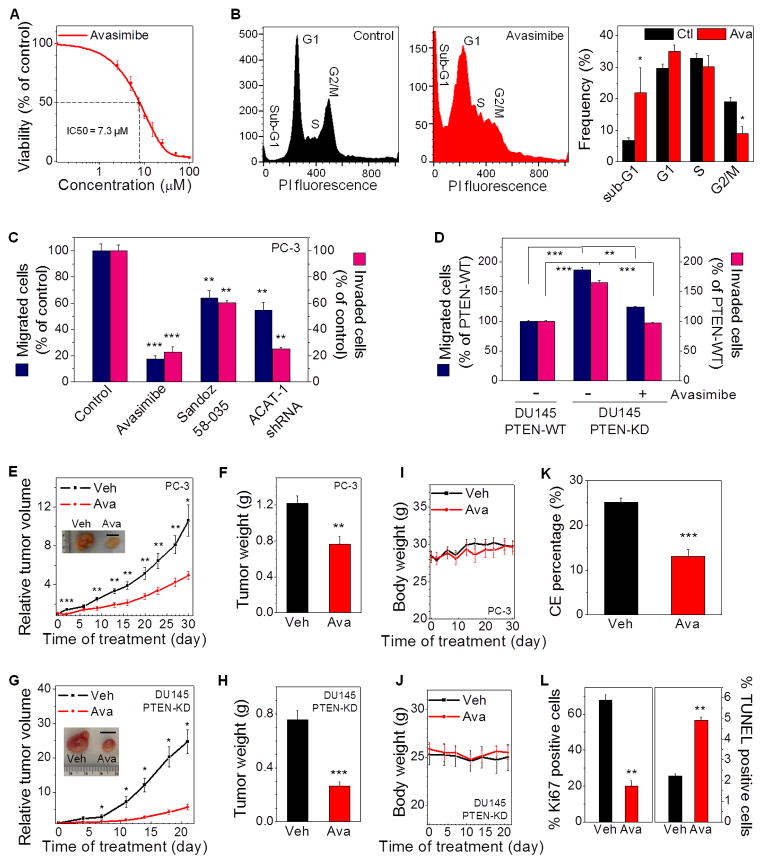
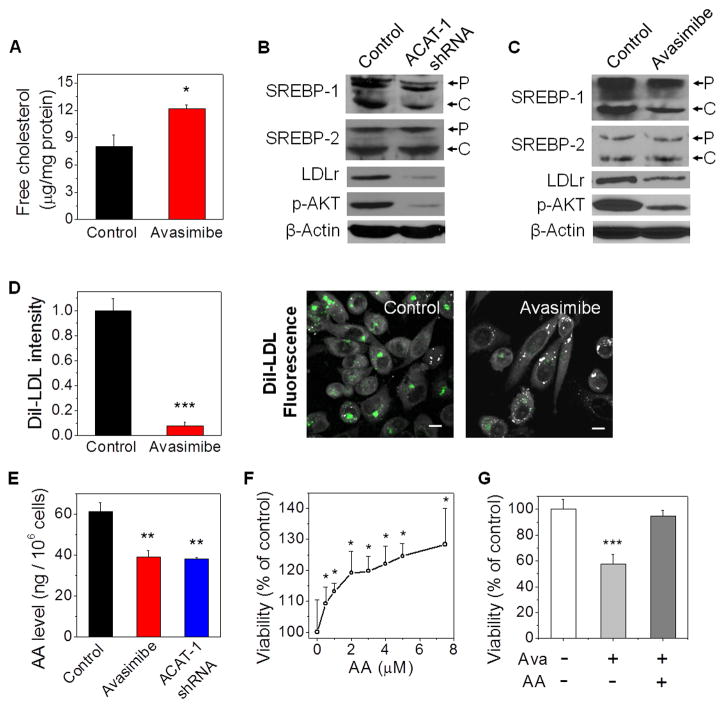
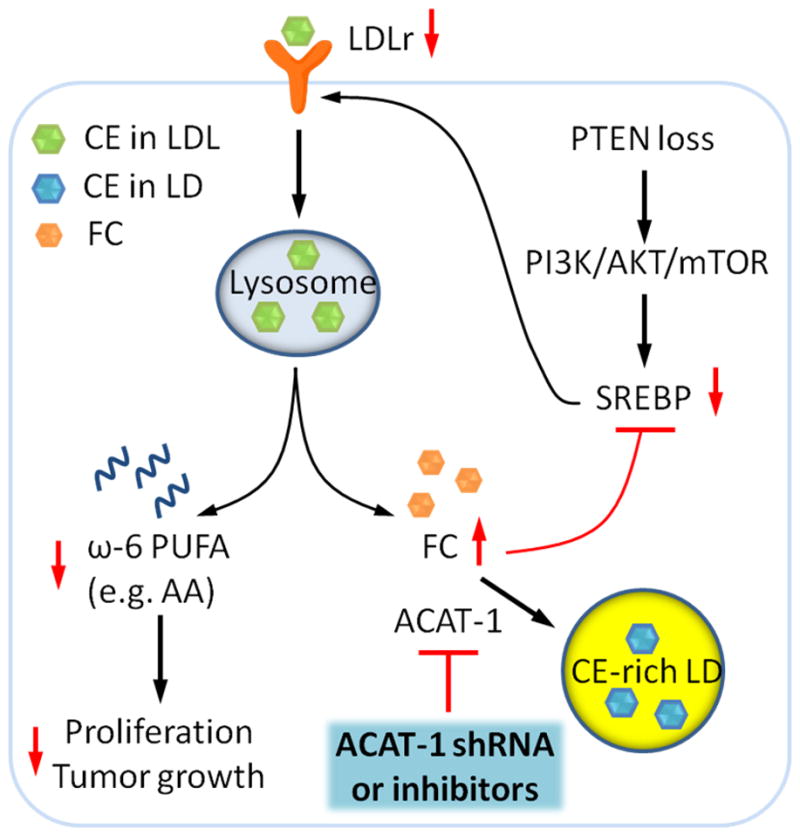
Altered lipid metabolism is increasingly recognized as a signature of cancer cells. Enabled by label-free Raman spectromicroscopy, we performed quantitative analysis of lipogenesis at single-cell level in human patient cancerous tissues. Our imaging data revealed an unexpected, aberrant accumulation of esterified cholesterol in lipid droplets of high-grade prostate cancer and metastases. Biochemical study showed that such cholesteryl ester accumulation was a consequence of loss of tumor suppressor PTEN and subsequent activation of PI3K/AKT pathway in prostate cancer cells. Furthermore, we found that such accumulation arose from significantly enhanced uptake of exogenous lipoproteins and required cholesterol esterification. Depletion of cholesteryl ester storage significantly reduced cancer proliferation, impaired cancer invasion capability, and suppressed tumor growth in mouse xenograft models with negligible toxicity. These findings open opportunities for diagnosing and treating prostate cancer by targeting the altered cholesterol metabolism.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Cholesteryl esters: fueling the fury of prostate cancer.Cell Metab. 2014 Mar 4;19(3):350-2. doi: 10.1016/j.cmet.2014.02.012. Cell Metab. 2014. PMID: 24606894
-
Lipogenic metabolism: a viable target for prostate cancer treatment?Asian J Androl. 2014 Sep-Oct;16(5):661-3. doi: 10.4103/1008-682X.132947. Asian J Androl. 2014. PMID: 24969061 Free PMC article.
References
-
- Ablin RJ, Guinan P, Bush IM. Lipofuscin granules in normal, benign and malignant human prostatic tissue. Urol Res. 1973;1:149–151. - PubMed
-
- Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, Viola JPB. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008;68:1732–1740. - PubMed
-
- Antalis C, Arnold T, Rasool T, Lee B, Buhman K, Siddiqui R. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res Treat. 2009;122:661–670. - PubMed
-
- Bemlih S, Poirier MD, Andaloussi AE. Acyl-coenzyme A: Cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol Ther. 2010;9:1025–1032. - PubMed
-
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
