

Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth
- PMID: 24607389
- PMCID: PMC3951929
- DOI: 10.1016/j.ajhg.2014.02.004
Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth
Abstract
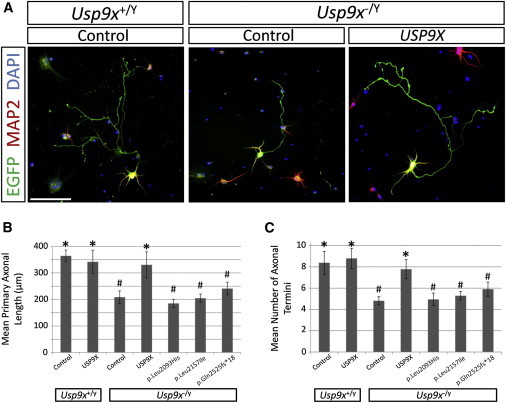
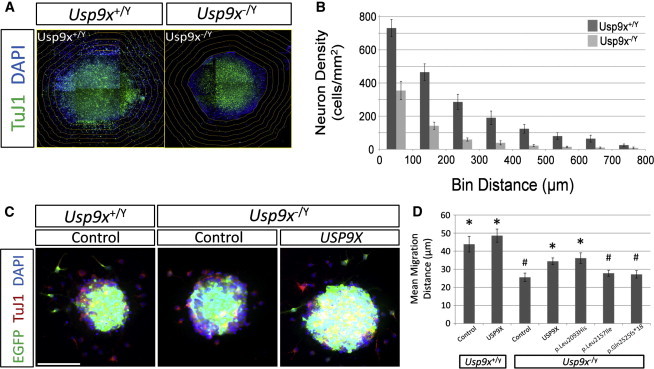
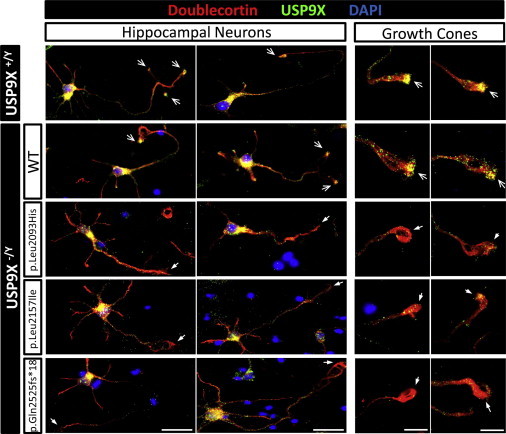
With a wealth of disease-associated DNA variants being recently reported, the challenges of providing their functional characterization are mounting. Previously, as part of a large systematic resequencing of the X chromosome in 208 unrelated families with nonsyndromic X-linked intellectual disability, we identified three unique variants (two missense and one protein truncating) in USP9X. To assess the functional significance of these variants, we took advantage of the Usp9x knockout mouse we generated. Loss of Usp9x causes reduction in both axonal growth and neuronal cell migration. Although overexpression of wild-type human USP9X rescued these defects, all three USP9X variants failed to rescue axonal growth, caused reduced USP9X protein localization in axonal growth cones, and (in 2/3 variants) failed to rescue neuronal cell migration. Interestingly, in one of these families, the proband was subsequently identified to have a microdeletion encompassing ARID1B, a known ID gene. Given our findings it is plausible that loss of function of both genes contributes to the individual's phenotype. This case highlights the complexity of the interpretations of genetic findings from genome-wide investigations. We also performed proteomics analysis of neurons from both the wild-type and Usp9x knockout embryos and identified disruption of the cytoskeleton as the main underlying consequence of the loss of Usp9x. Detailed clinical assessment of all three families with USP9X variants identified hypotonia and behavioral and morphological defects as common features in addition to ID. Together our data support involvement of all three USP9X variants in ID in these families and provide likely cellular and molecular mechanisms involved.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
