

Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats
- PMID: 24608905
- PMCID: PMC3946746
- DOI: 10.1371/journal.pone.0091253
Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats
Abstract
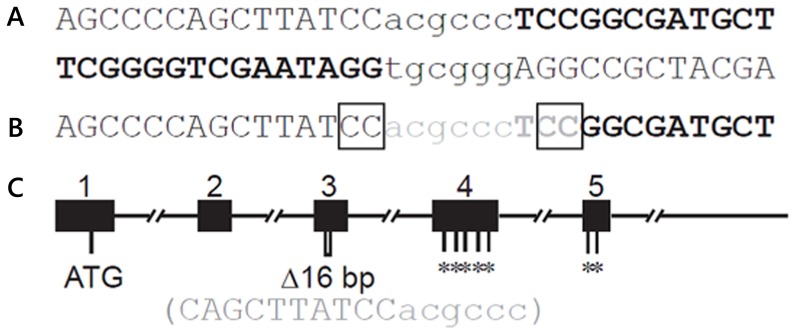
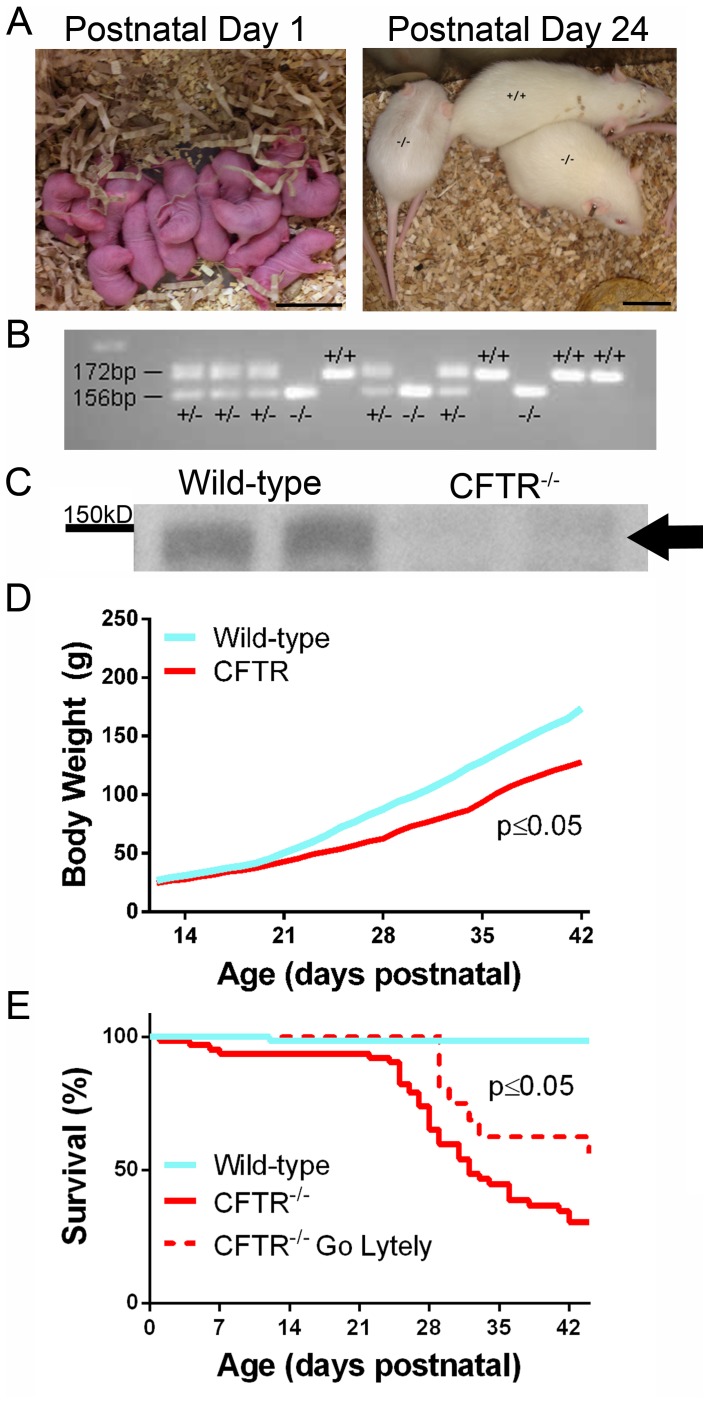
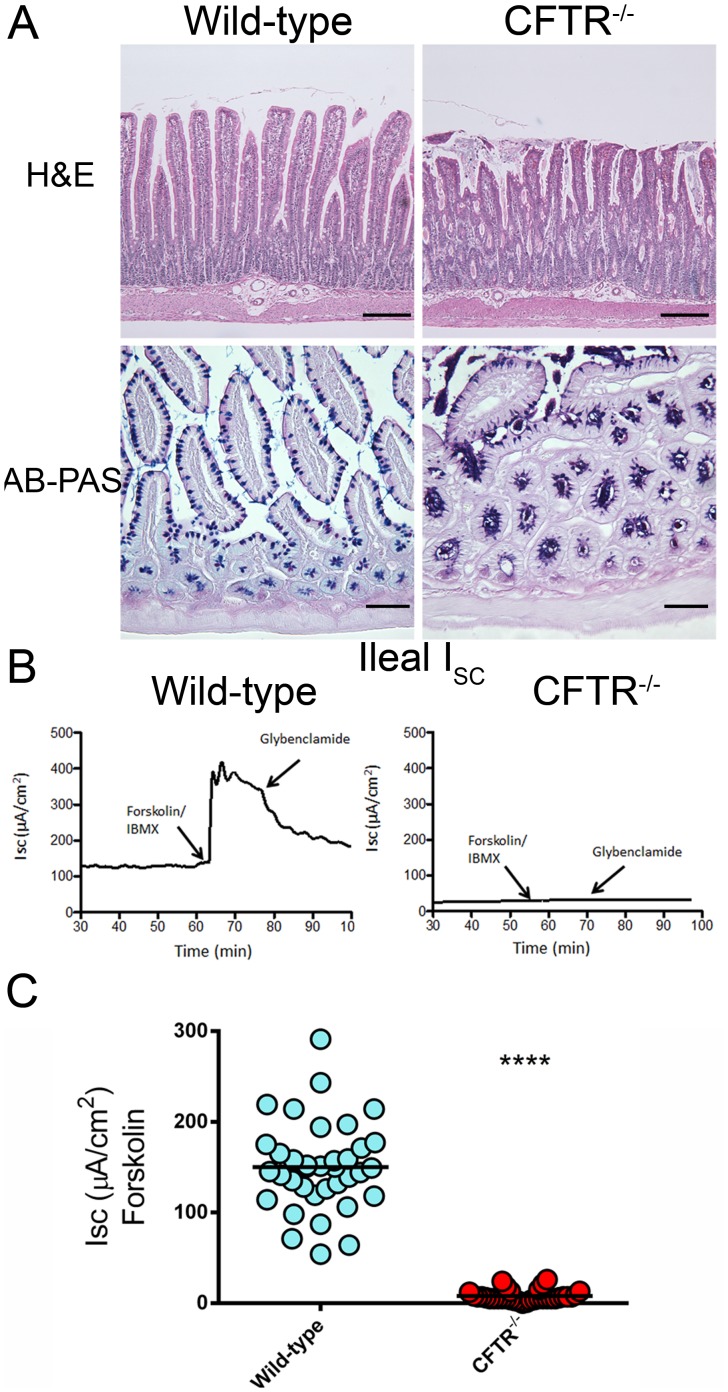
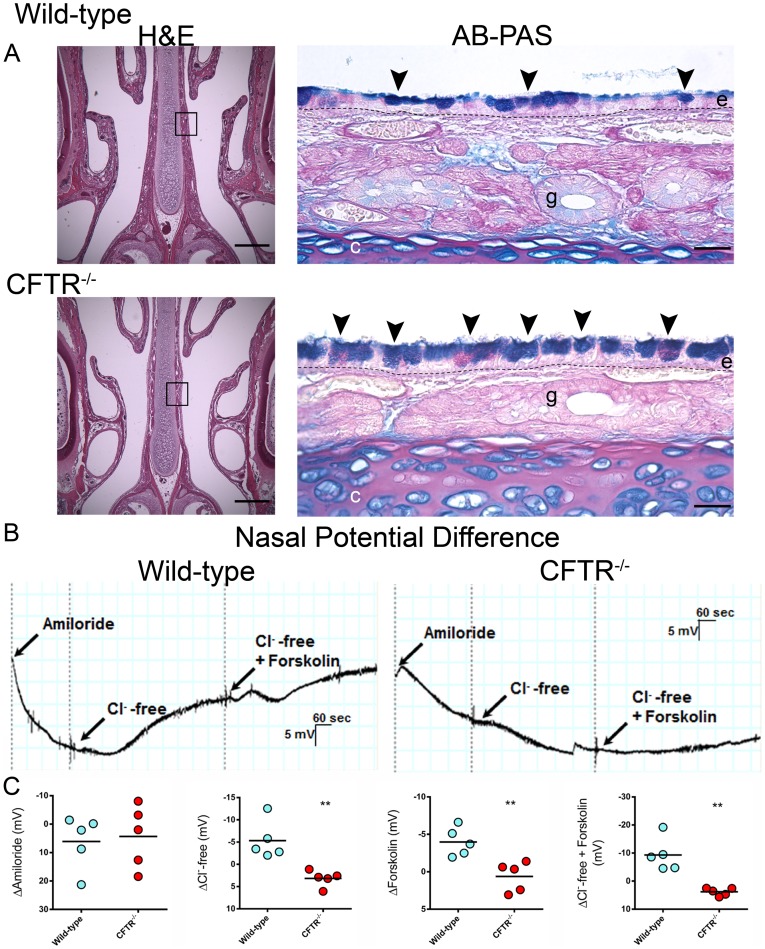
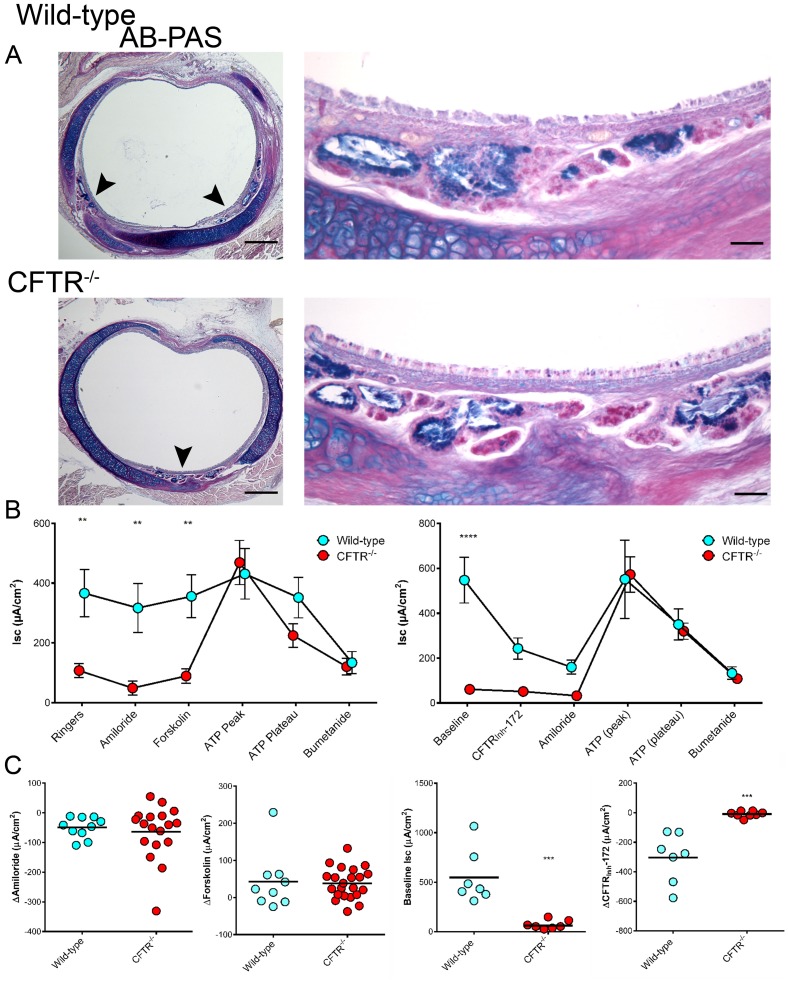
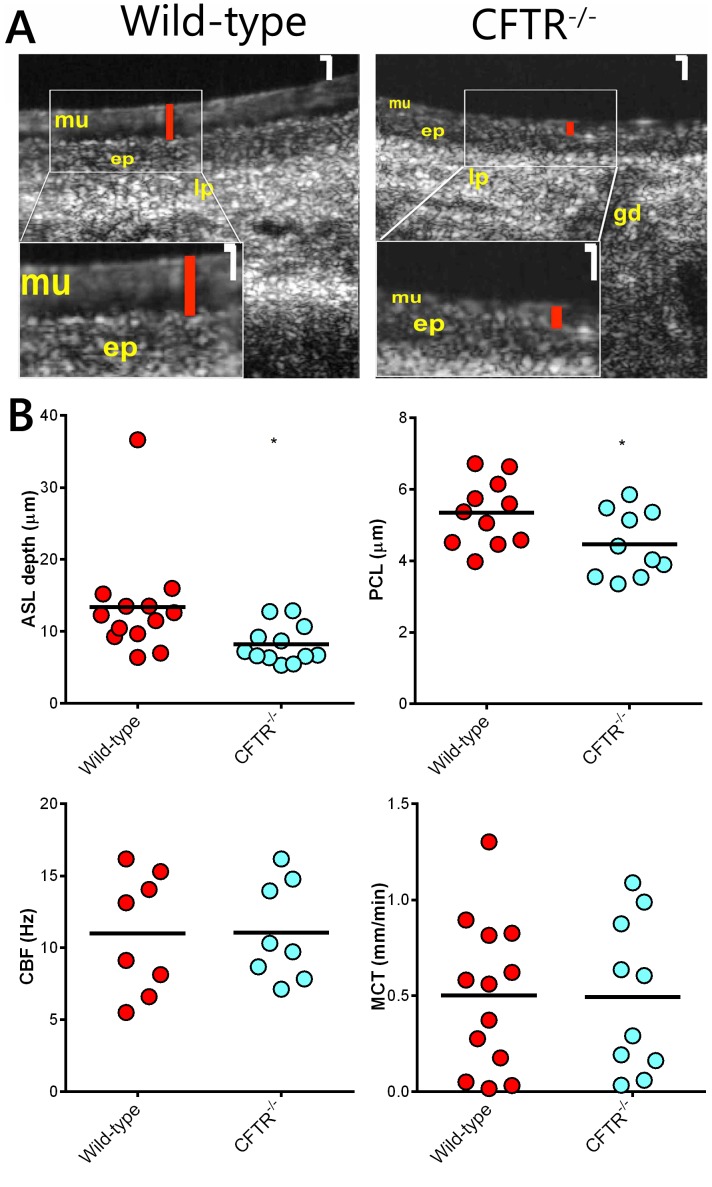
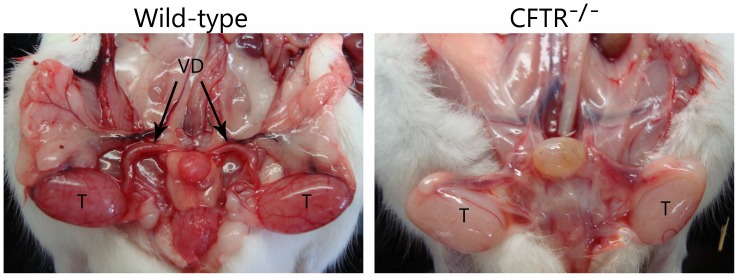
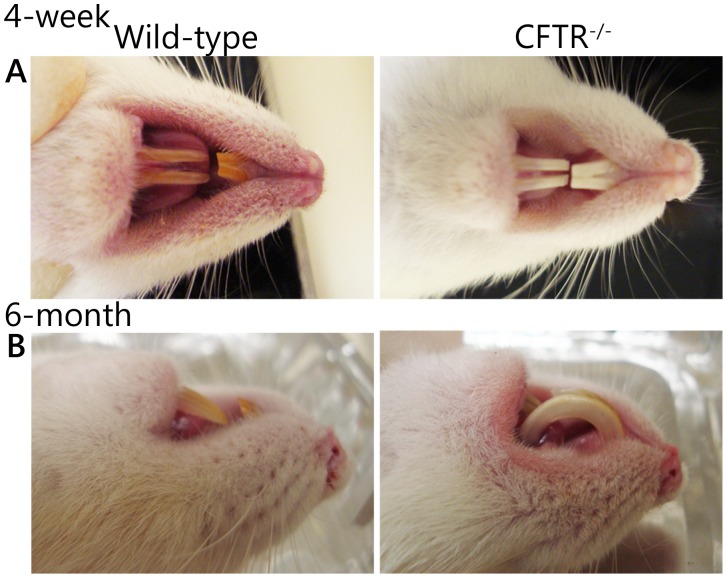
Animal models for cystic fibrosis (CF) have contributed significantly to our understanding of disease pathogenesis. Here we describe development and characterization of the first cystic fibrosis rat, in which the cystic fibrosis transmembrane conductance regulator gene (CFTR) was knocked out using a pair of zinc finger endonucleases (ZFN). The disrupted Cftr gene carries a 16 base pair deletion in exon 3, resulting in loss of CFTR protein expression. Breeding of heterozygous (CFTR+/-) rats resulted in Mendelian distribution of wild-type, heterozygous, and homozygous (CFTR-/-) pups. Nasal potential difference and transepithelial short circuit current measurements established a robust CF bioelectric phenotype, similar in many respects to that seen in CF patients. Young CFTR-/- rats exhibited histological abnormalities in the ileum and increased intracellular mucus in the proximal nasal septa. By six weeks of age, CFTR-/- males lacked the vas deferens bilaterally. Airway surface liquid and periciliary liquid depth were reduced, and submucosal gland size was abnormal in CFTR-/- animals. Use of ZFN based gene disruption successfully generated a CF animal model that recapitulates many aspects of human disease, and may be useful for modeling other CF genotypes, including CFTR processing defects, premature truncation alleles, and channel gating abnormalities.
Conflict of interest statement
Figures
References
-
- Grosse SD, Boyle CA, Botkin JR, Comeau AM, Kharrazi M, et al. (2004) Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep 53: 1–36. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073. - PubMed
-
- Guilbault C, Saeed Z, Downey GP, Radzioch D (2007) Cystic fibrosis mouse models. Am J Respir Cell Mol Biol 36: 1–7. - PubMed
-
- Carvalho-Oliveira I, Scholte BJ, Penque D (2007) What have we learned from mouse models for cystic fibrosis? Expert Rev Mol Diagn 7: 407–417. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
