

Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity
- PMID: 24613410
- PMCID: PMC4048950
- DOI: 10.1016/j.cellsig.2014.02.017
Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity
Abstract
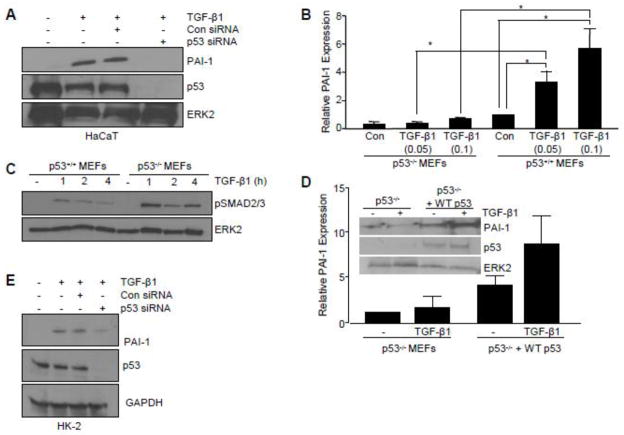
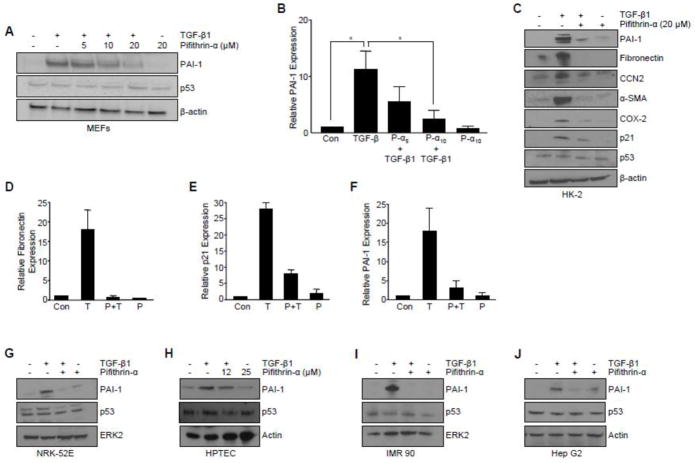
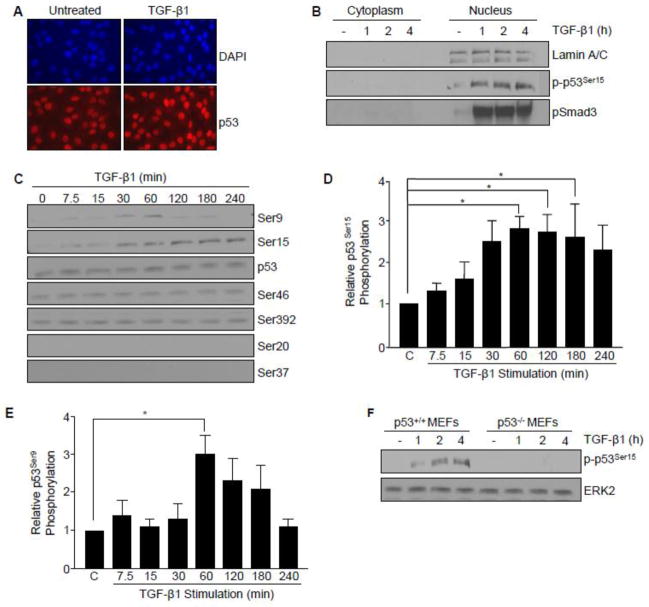
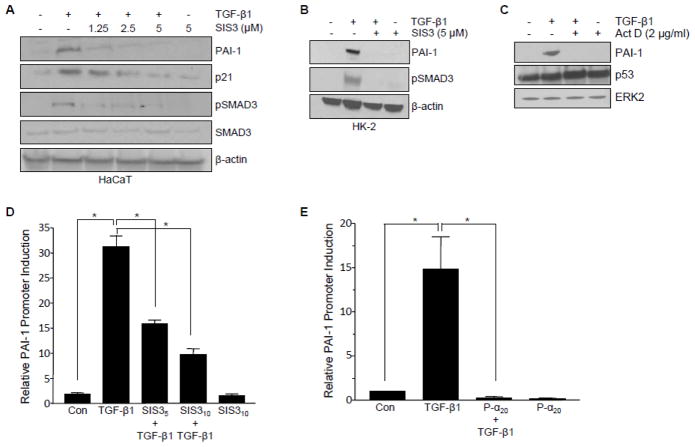
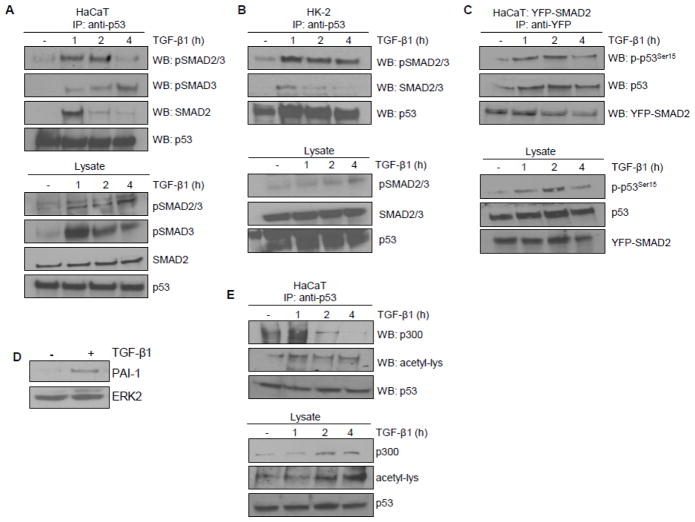
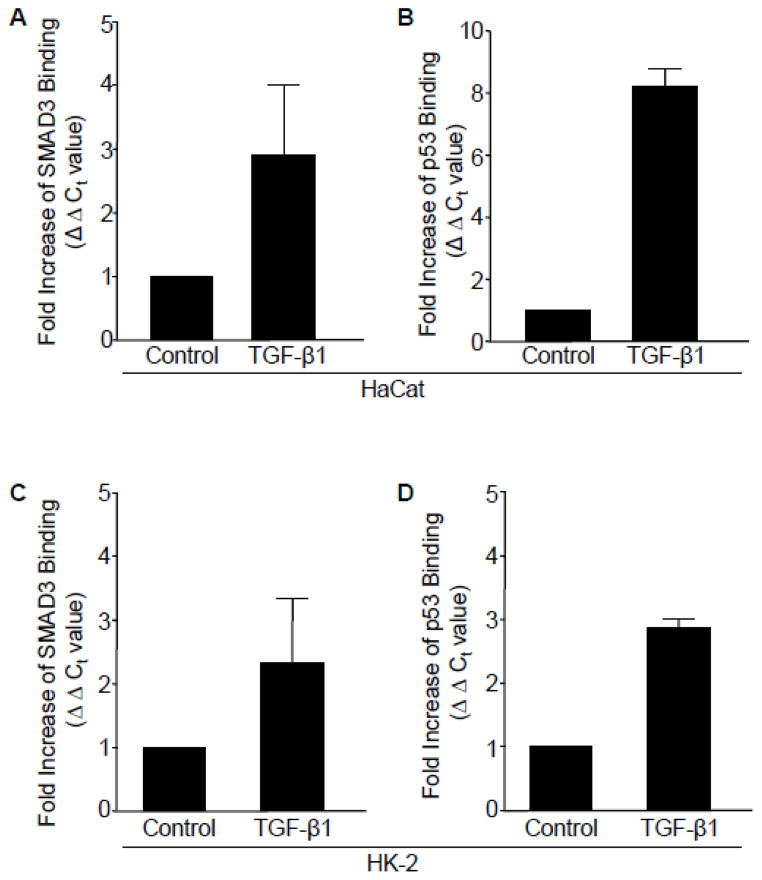
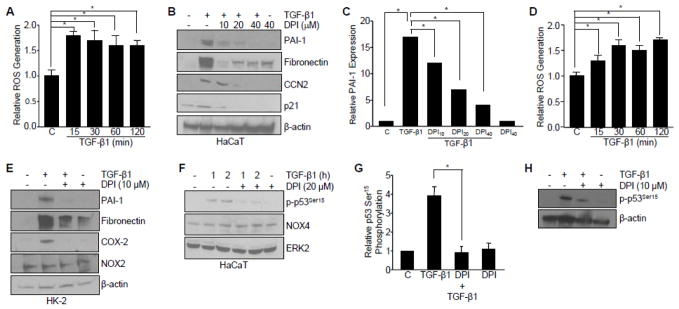
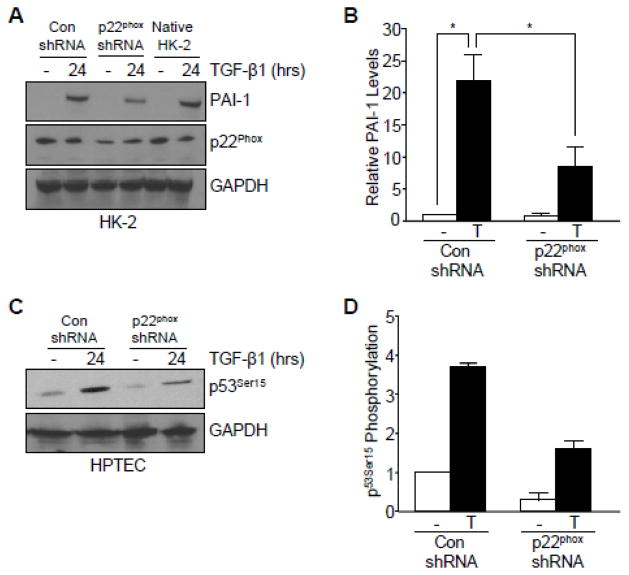
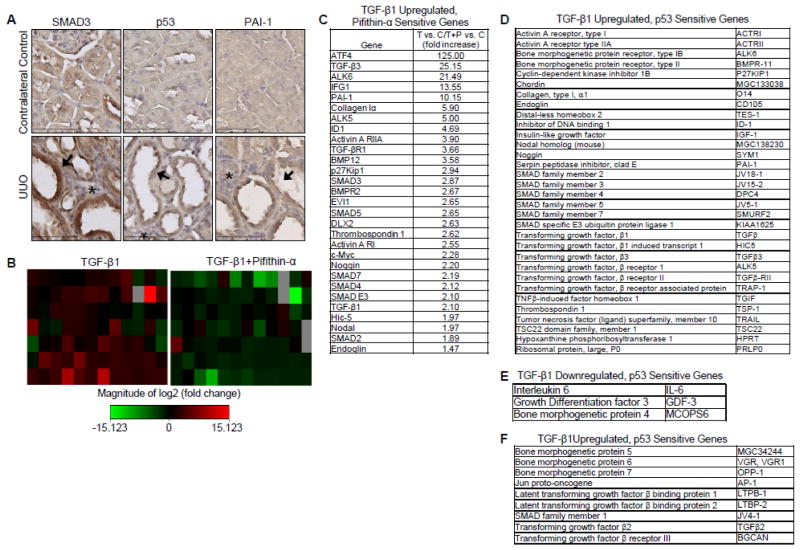
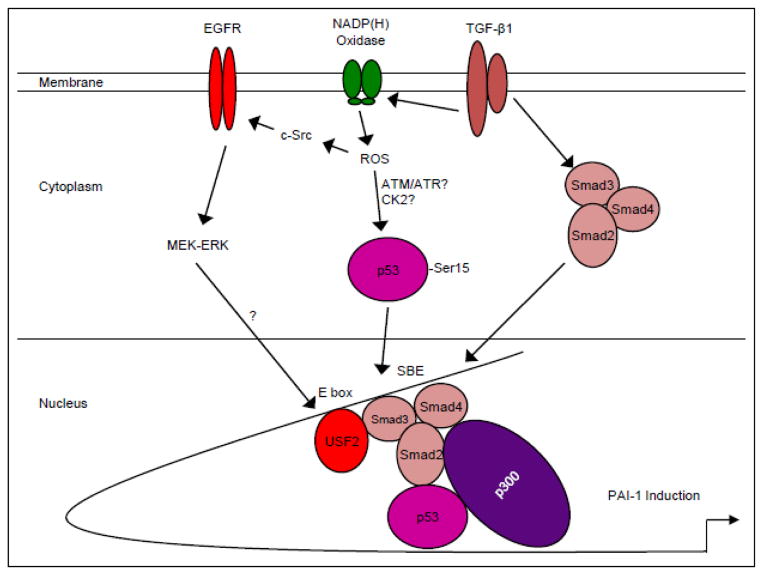
Transforming growth factor-β1 (TGF-β1) regulates the tissue response to injury and is the principal driver of excessive scarring leading to fibrosis and eventual organ failure. The TGF-β1 effectors SMAD3 and p53 are major contributors to disease progression. While SMAD3 is an established pro-fibrotic factor, the role of p53 in the TGF-β1-induced fibrotic program is not clear. p53 gene silencing, genetic ablation/subsequent rescue, and pharmacological inhibition confirmed that p53 was required for expression of plasminogen activator inhibitor-1 (PAI-1), a major TGF-β1 target gene and a key causative element in fibrotic disorders. TGF-β1 regulated p53 activity by stimulating p53(Ser15 and 9) phosphorylation and acetylation, promoting interactions with activated SMADs and subsequent binding of p53/SMAD3 to the PAI-1 promoter in HK-2 human renal tubular epithelial cells and HaCaT human keratinocytes. Immunohistochemistry revealed prominent co-induction of SMAD3, p53 and PAI-1 in the tubular epithelium of the obstructed kidney consistent with a potential in vivo role for p53 and SMADs in TGF-β1-driven renal fibrosis. TGF-β1-initiated phosphorylation of p53(Ser15) and up-regulation of expression of several pro-fibrotic genes, moreover, was dependent on the rapid generation of reactive oxygen species (ROS). shRNA silencing of the p22(Phox) subunit of NADP(H) oxidases in HK-2 cells partially attenuated (over 50%) p53(Ser15) phosphorylation and PAI-1 induction. These studies highlight the role of free radicals in p53 activation and subsequent pro-fibrotic reprogramming by TGF-β1 via the SMAD3-p53 transcriptional axis. Present findings provide a rationale for therapeutic targeting of SMAD3-p53 in aberrant TGF-β1 signaling associated with renal fibrosis.
Keywords: Chromatin immunoprecipitation; Gene expression; PAI-1; Reactive oxygen species; SMADs; TGF-β1; Tissue fibrosis; Transcription; p53.
Copyright © 2014 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have no conflict of interest.
Figures
Similar articles
-
Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species.Cell Signal. 2013 Nov;25(11):2198-209. doi: 10.1016/j.cellsig.2013.07.007. Epub 2013 Jul 18. Cell Signal. 2013. PMID: 23872073
-
Tumor suppressor ataxia telangiectasia mutated functions downstream of TGF-β1 in orchestrating profibrotic responses.FASEB J. 2015 Apr;29(4):1258-68. doi: 10.1096/fj.14-262527. Epub 2014 Dec 5. FASEB J. 2015. PMID: 25480384 Free PMC article.
-
Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways.FASEB J. 2019 Sep;33(9):9797-9810. doi: 10.1096/fj.201802489RR. Epub 2019 May 16. FASEB J. 2019. PMID: 31095421 Free PMC article.
-
TGF-β1-p53 cooperativity regulates a profibrotic genomic program in the kidney: molecular mechanisms and clinical implications.FASEB J. 2019 Oct;33(10):10596-10606. doi: 10.1096/fj.201900943R. Epub 2019 Jul 6. FASEB J. 2019. PMID: 31284746 Free PMC article. Review.
-
TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities.Cell Signal. 2013 Jan;25(1):264-8. doi: 10.1016/j.cellsig.2012.10.003. Epub 2012 Oct 11. Cell Signal. 2013. PMID: 23063463 Free PMC article. Review.
Cited by
-
Accredited translational medicine centre: Human renal fibrotic disease: Translational research at the Center for Cell Biology and Cancer Research (CCBCR), Albany Medical College, Albany, NY.New Horiz Transl Med. 2015 Jan;2(2):51-54. doi: 10.1016/j.nhtm.2014.12.001. New Horiz Transl Med. 2015. PMID: 25692175 Free PMC article. No abstract available.
-
Acute Kidney Injury and Progression of Diabetic Kidney Disease.Adv Chronic Kidney Dis. 2018 Mar;25(2):166-180. doi: 10.1053/j.ackd.2017.12.005. Adv Chronic Kidney Dis. 2018. PMID: 29580581 Free PMC article. Review.
-
Dapagliflozin can alleviate renal fibrosis in rats with streptozotocin‑induced type 2 diabetes mellitus.Exp Ther Med. 2023 Oct 24;26(6):572. doi: 10.3892/etm.2023.12271. eCollection 2023 Dec. Exp Ther Med. 2023. PMID: 38023356 Free PMC article.
-
A novel role of kallikrein-related peptidase 8 in the pathogenesis of diabetic cardiac fibrosis.Theranostics. 2021 Feb 20;11(9):4207-4231. doi: 10.7150/thno.48530. eCollection 2021. Theranostics. 2021. PMID: 33754057 Free PMC article.
-
The aryl hydrocarbon receptor agonist benzo(a)pyrene reactivates LINE-1 in HepG2 cells through canonical TGF-β1 signaling: implications in hepatocellular carcinogenesis.Am J Cancer Res. 2016 May 1;6(5):1066-77. eCollection 2016. Am J Cancer Res. 2016. PMID: 27293999 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
