

Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor β1 signaling in metastatic breast cancers
- PMID: 24618085
- PMCID: PMC4053226
- DOI: 10.1186/bcr3623
Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor β1 signaling in metastatic breast cancers
Abstract
Introduction: Epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET) facilitate breast cancer (BC) metastasis; however, stable molecular changes that result as a consequence of these processes remain poorly defined. Therefore, with the hope of targeting unique aspects of metastatic tumor outgrowth, we sought to identify molecular markers that could identify tumor cells that had completed the EMT:MET cycle.
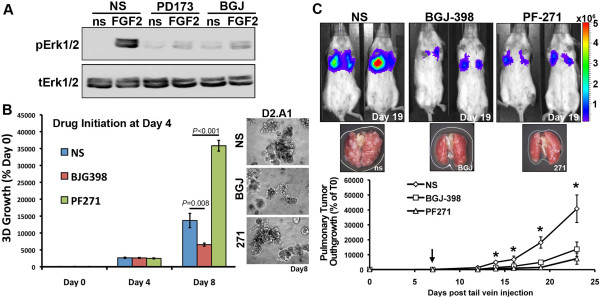
Methods: An in vivo reporter system for epithelial cadherin (E-cad) expression was used to quantify its regulation in metastatic BC cells during primary and metastatic tumor growth. Exogenous addition of transforming growth factor β1 (TGF-β1) was used to induce EMT in an in situ model of BC. Microarray analysis was employed to examine gene expression changes in cells chronically treated with and withdrawn from TGF-β1, thus completing one full EMT:MET cycle. Changes in fibroblast growth factor receptor type 1 (FGFR1) isoform expression were validated using PCR analyses of patient-derived tumor tissues versus matched normal tissues. FGFR1 gene expression was manipulated using short hairpin RNA depletion and cDNA rescue. Preclinical pharmacological inhibition of FGFR kinase was employed using the orally available compound BGJ-398.
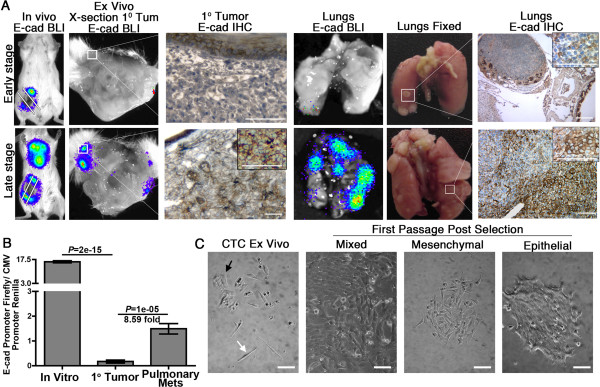
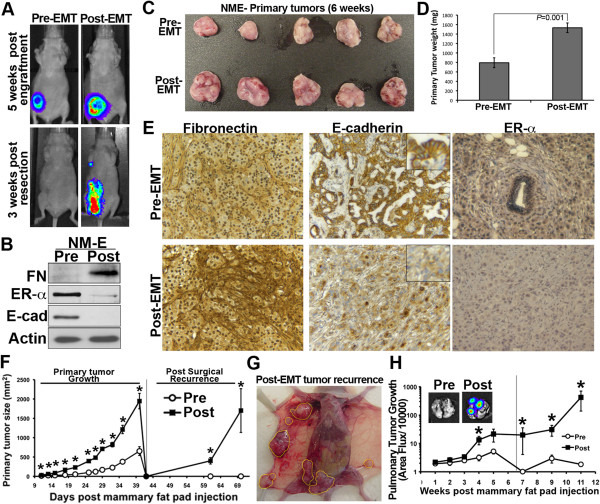
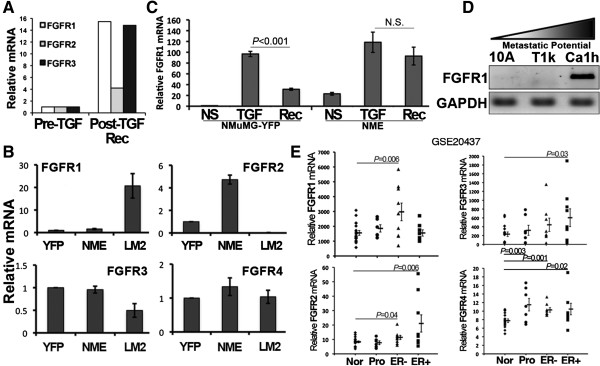
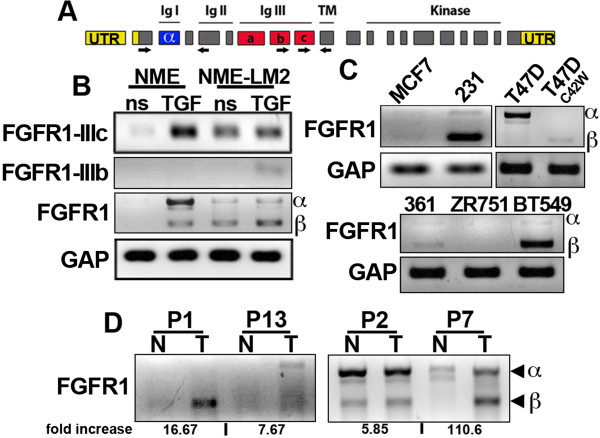
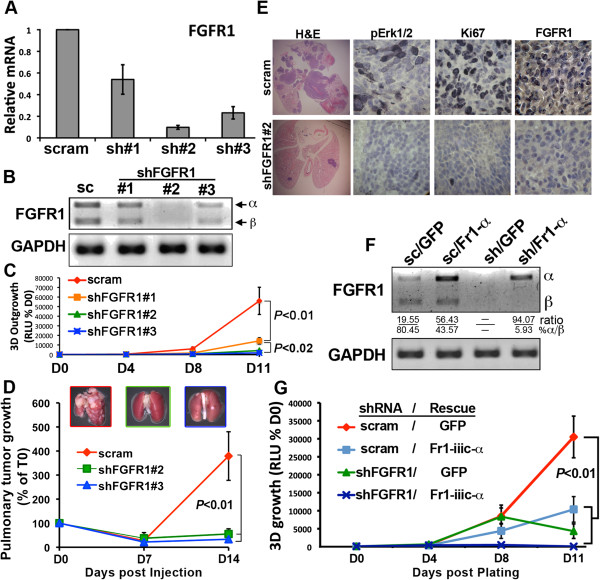
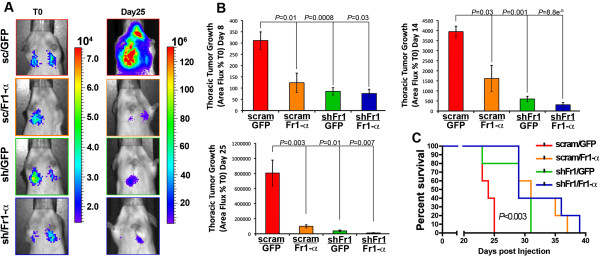
Results: Metastatic BC cells undergo spontaneous downregulation of E-cad during primary tumor growth, and its expression subsequently returns following initiation of metastatic outgrowth. Exogenous exposure to TGF-β1 was sufficient to drive the metastasis of an otherwise in situ model of BC and was similarly associated with a depletion and return of E-cad expression during metastatic progression. BC cells treated and withdrawn from TGF-β stably upregulate a truncated FGFR1-β splice variant that lacks the outermost extracellular immunoglobulin domain. Identification of this FGFR1 splice variant was verified in metastatic human BC cell lines and patient-derived tumor samples. Expression of FGFR1-β was also dominant in a model of metastatic outgrowth where depletion of FGFR1 and pharmacologic inhibition of FGFR kinase activity both inhibited pulmonary tumor outgrowth. Highlighting the dichotomous nature of FGFR splice variants and recombinant expression of full-length FGFR1-α also blocked pulmonary tumor outgrowth.
Conclusion: The results of our study strongly suggest that FGFR1-β is required for the pulmonary outgrowth of metastatic BC. Moreover, FGFR1 isoform expression can be used as a predictive biomarker for therapeutic application of its kinase inhibitors.
Figures
Similar articles
-
TGF-β stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer.Oncogene. 2013 Apr 18;32(16):2005-15. doi: 10.1038/onc.2012.230. Epub 2012 Jun 18. Oncogene. 2013. PMID: 22710711 Free PMC article.
-
Epigenetic targeting of neuropilin-1 prevents bypass signaling in drug-resistant breast cancer.Oncogene. 2021 Jan;40(2):322-333. doi: 10.1038/s41388-020-01530-6. Epub 2020 Oct 30. Oncogene. 2021. PMID: 33128042 Free PMC article.
-
Covalent Targeting of Fibroblast Growth Factor Receptor Inhibits Metastatic Breast Cancer.Mol Cancer Ther. 2016 Sep;15(9):2096-106. doi: 10.1158/1535-7163.MCT-16-0136. Epub 2016 Jul 1. Mol Cancer Ther. 2016. PMID: 27371729 Free PMC article.
-
The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells.J Mammary Gland Biol Neoplasia. 2010 Jun;15(2):169-90. doi: 10.1007/s10911-010-9181-1. Epub 2010 May 15. J Mammary Gland Biol Neoplasia. 2010. PMID: 20467795 Free PMC article. Review.
-
Noncanonical TGF-β signaling during mammary tumorigenesis.J Mammary Gland Biol Neoplasia. 2011 Jun;16(2):127-46. doi: 10.1007/s10911-011-9207-3. Epub 2011 Mar 31. J Mammary Gland Biol Neoplasia. 2011. PMID: 21448580 Free PMC article. Review.
Cited by
-
MRI detection of breast cancer micrometastases with a fibronectin-targeting contrast agent.Nat Commun. 2015 Aug 12;6:7984. doi: 10.1038/ncomms8984. Nat Commun. 2015. PMID: 26264658 Free PMC article.
-
Regulation of epithelial-mesenchymal transition and metastasis by TGF-β, P-bodies, and autophagy.Oncotarget. 2017 Oct 17;8(61):103302-103314. doi: 10.18632/oncotarget.21871. eCollection 2017 Nov 28. Oncotarget. 2017. PMID: 29262563 Free PMC article.
-
Computational Inferences of the Functions of Alternative/Noncanonical Splice Isoforms Specific to HER2+/ER-/PR- Breast Cancers, a Chromosome 17 C-HPP Study.J Proteome Res. 2015 Sep 4;14(9):3519-29. doi: 10.1021/acs.jproteome.5b00498. Epub 2015 Jul 23. J Proteome Res. 2015. PMID: 26147891 Free PMC article.
-
Pharmacologic Inhibition of FGFR Modulates the Metastatic Immune Microenvironment and Promotes Response to Immune Checkpoint Blockade.Cancer Immunol Res. 2020 Dec;8(12):1542-1553. doi: 10.1158/2326-6066.CIR-20-0235. Epub 2020 Oct 22. Cancer Immunol Res. 2020. PMID: 33093218 Free PMC article.
-
FGFR1 is a potential therapeutic target in neuroblastoma.Cancer Cell Int. 2022 Apr 29;22(1):174. doi: 10.1186/s12935-022-02587-x. Cancer Cell Int. 2022. PMID: 35488346 Free PMC article.
References
-
- Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y. Direct targeting of Sec23a by miR-200 s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–1108. doi: 10.1038/nm.2401. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
