

Proteomics in heart failure: top-down or bottom-up?
- PMID: 24619480
- PMCID: PMC4037365
- DOI: 10.1007/s00424-014-1471-9
Proteomics in heart failure: top-down or bottom-up?
Abstract
The pathophysiology of heart failure (HF) is diverse, owing to multiple etiologies and aberrations in a number of cellular processes. Therefore, it is essential to understand how defects in the molecular pathways that mediate cellular responses to internal and external stressors function as a system to drive the HF phenotype. Mass spectrometry (MS)-based proteomics strategies have great potential for advancing our understanding of disease mechanisms at the systems level because proteins are the effector molecules for all cell functions and, thus, are directly responsible for determining cell phenotype. Two MS-based proteomics strategies exist: peptide-based bottom-up and protein-based top-down proteomics--each with its own unique strengths and weaknesses for interrogating the proteome. In this review, we will discuss the advantages and disadvantages of bottom-up and top-down MS for protein identification, quantification, and analysis of post-translational modifications, as well as highlight how both of these strategies have contributed to our understanding of the molecular and cellular mechanisms underlying HF. Additionally, the challenges associated with both proteomics approaches will be discussed and insights will be offered regarding the future of MS-based proteomics in HF research.
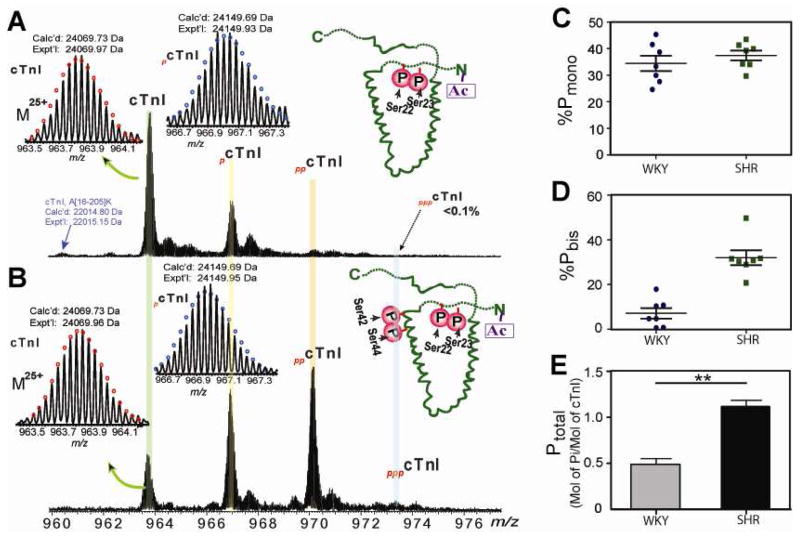
Figures

References
-
- Addona TA, Shi X, Keshishian H, Mani DR, Burgess M, Gillette MA, Clauser KR, Shen D, Lewis GD, Farrell LA, Fifer MA, Sabatine MS, Gerszten RE, Carr SA. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat Biotechnol. 2011;29:635–43. - PMC - PubMed
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. - PMC - PubMed
-
- Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc Natl Acad Sci U S A. 2013;110:10153–8. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
