

When heat casts a spell on the DNA damage checkpoints
- PMID: 24621867
- PMCID: PMC3971410
- DOI: 10.1098/rsob.140008
When heat casts a spell on the DNA damage checkpoints
Abstract
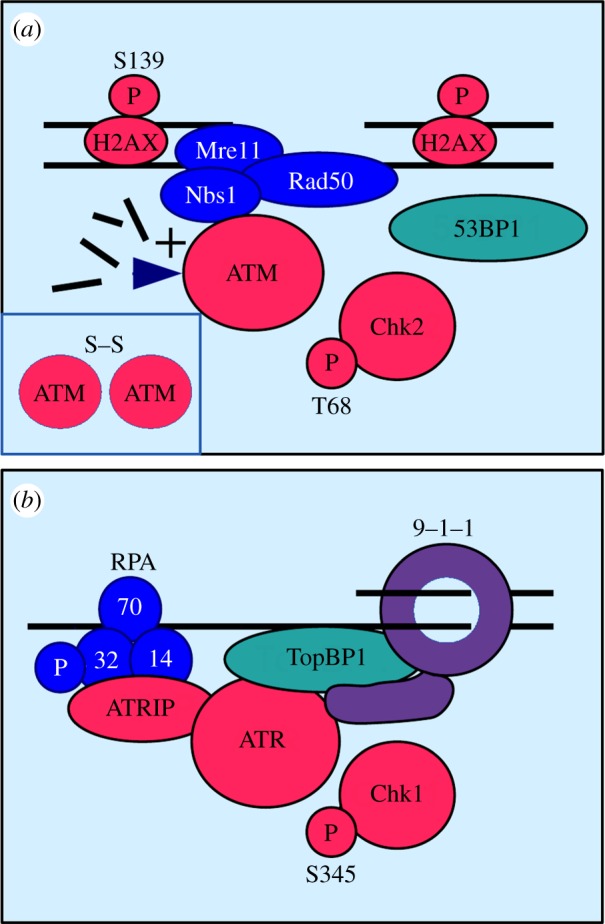
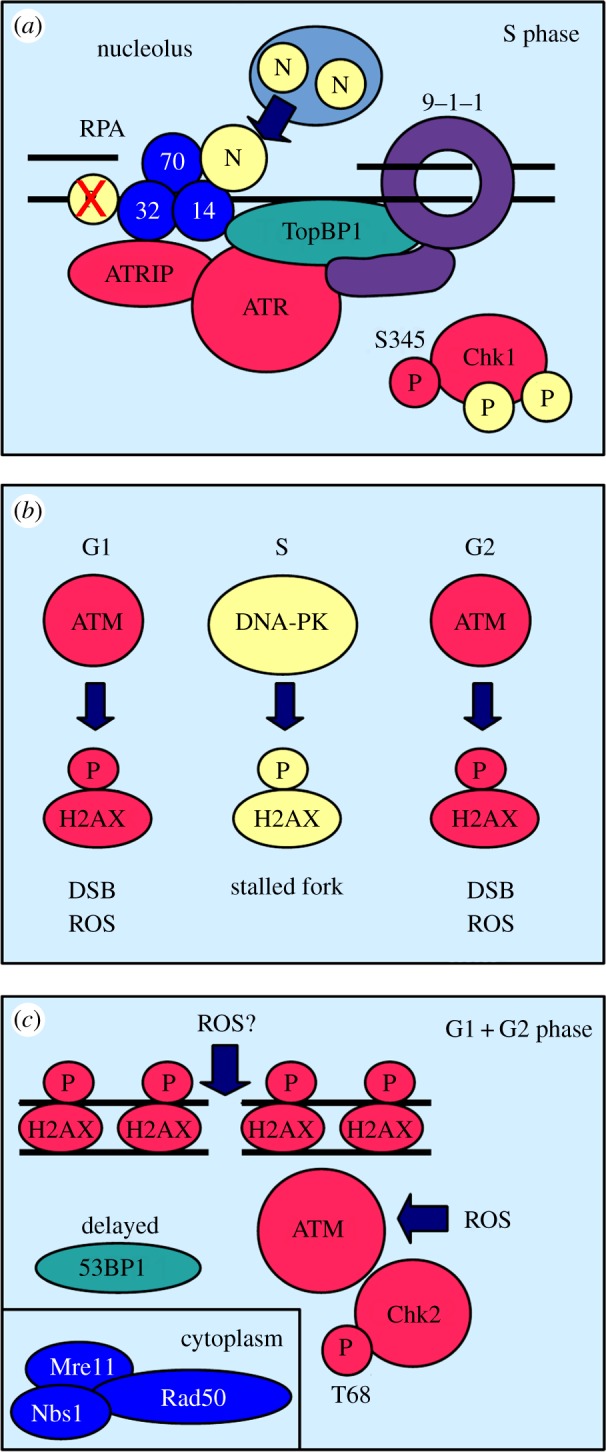
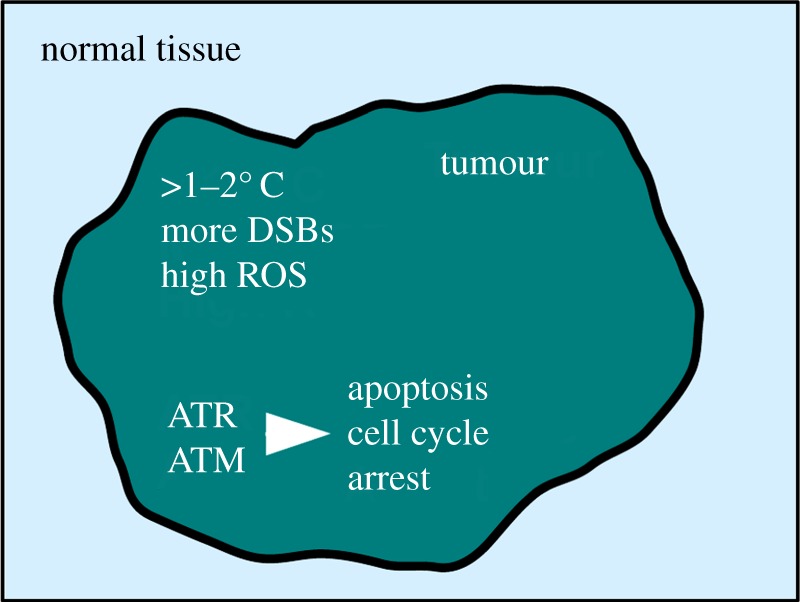
Peregrine Laziosi (1265-1345), an Italian priest, became the patron saint of cancer patients when the tumour in his left leg miraculously disappeared after he developed a fever. Elevated body temperature can cause tumours to regress and sensitizes cancer cells to agents that break DNA. Why hyperthermia blocks the repair of broken chromosomes by changing the way that the DNA damage checkpoint kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) are activated is an unanswered question. This review discusses the current knowledge of how heat affects the ATR-Chk1 and ATM-Chk2 kinase networks, and provides a possible explanation of why homeothermal organisms such as humans still possess this ancient heat response.
Keywords: ATM; ATR; Chk1; Chk2; DNA damage checkpoint; hyperthermia.
Figures
Similar articles
-
Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage.J Biol Chem. 2005 Jan 14;280(2):1186-92. doi: 10.1074/jbc.M410873200. Epub 2004 Nov 8. J Biol Chem. 2005. PMID: 15533933
-
Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation.J Biol Chem. 2003 Apr 25;278(17):14806-11. doi: 10.1074/jbc.M210862200. Epub 2003 Feb 14. J Biol Chem. 2003. PMID: 12588868
-
DNA damage checkpoint kinases in cancer.Expert Rev Mol Med. 2020 Jun 8;22:e2. doi: 10.1017/erm.2020.3. Expert Rev Mol Med. 2020. PMID: 32508294 Review.
-
N-nitroso-N-ethylurea activates DNA damage surveillance pathways and induces transformation in mammalian cells.BMC Cancer. 2014 Apr 24;14:287. doi: 10.1186/1471-2407-14-287. BMC Cancer. 2014. PMID: 24758542 Free PMC article.
-
Cancer Vulnerabilities Through Targeting the ATR/Chk1 and ATM/Chk2 Axes in the Context of DNA Damage.Cells. 2025 May 20;14(10):748. doi: 10.3390/cells14100748. Cells. 2025. PMID: 40422251 Free PMC article. Review.
Cited by
-
DNA Damage Repair in the Context of Plant Chromatin.Plant Physiol. 2015 Aug;168(4):1206-18. doi: 10.1104/pp.15.00538. Epub 2015 Jun 18. Plant Physiol. 2015. PMID: 26089404 Free PMC article. Review.
-
Scorpins in the DNA Damage Response.Int J Mol Sci. 2018 Jun 17;19(6):1794. doi: 10.3390/ijms19061794. Int J Mol Sci. 2018. PMID: 29914204 Free PMC article. Review.
-
p53 dynamics in single cells are temperature-sensitive.Sci Rep. 2020 Jan 30;10(1):1481. doi: 10.1038/s41598-020-58267-1. Sci Rep. 2020. PMID: 32001771 Free PMC article.
-
Emerging roles of intratumor microbiota in cancer: tumorigenesis and management strategies.J Transl Med. 2024 Sep 11;22(1):837. doi: 10.1186/s12967-024-05640-7. J Transl Med. 2024. PMID: 39261861 Free PMC article. Review.
-
The Topoisomerase 1 Inhibitor Austrobailignan-1 Isolated from Koelreuteria henryi Induces a G2/M-Phase Arrest and Cell Death Independently of p53 in Non-Small Cell Lung Cancer Cells.PLoS One. 2015 Jul 6;10(7):e0132052. doi: 10.1371/journal.pone.0132052. eCollection 2015. PLoS One. 2015. PMID: 26147394 Free PMC article.
References
-
- Bhuyan BK. 1979. Kinetics of cell kill by hyperthermia. Cancer Res 39, 2277–2284. - PubMed
-
- Saladino C, Ben-Hur E. 1976. Heat-enhanced radioresponse in HeLa cells. Isr. J. Med. Sci. 12, 134–138. - PubMed
-
- Corry PM, Robinson S, Getz S. 1977. Hyperthermic effects on DNA repair mechanisms. Radiology 123, 475–482. - PubMed
-
- Hehr T, Wust P, Bamberg M, Budach W. 2003. Current and potential role of thermoradiotherapy for solid tumours. Onkologie 26, 295–302. (doi:10.1159/000071628) - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
