

GABRA1 and STXBP1: novel genetic causes of Dravet syndrome
- PMID: 24623842
- PMCID: PMC4001207
- DOI: 10.1212/WNL.0000000000000291
GABRA1 and STXBP1: novel genetic causes of Dravet syndrome
Abstract
Objective: To determine the genes underlying Dravet syndrome in patients who do not have an SCN1A mutation on routine testing.
Methods: We performed whole-exome sequencing in 13 SCN1A-negative patients with Dravet syndrome and targeted resequencing in 67 additional patients to identify new genes for this disorder.
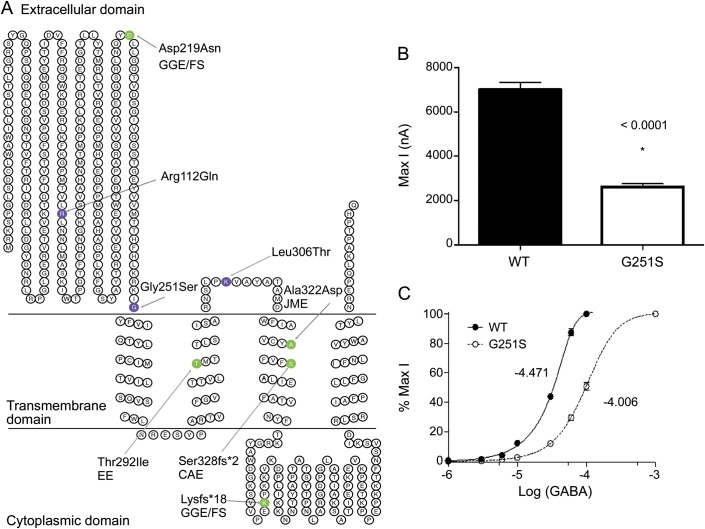
Results: We detected disease-causing mutations in 2 novel genes for Dravet syndrome, with mutations in GABRA1 in 4 cases and STXBP1 in 3. Furthermore, we identified 3 patients with previously undetected SCN1A mutations, suggesting that SCN1A mutations occur in even more than the currently accepted ∼ 75% of cases.
Conclusions: We show that GABRA1 and STXBP1 make a significant contribution to Dravet syndrome after SCN1A abnormalities have been excluded. Our results have important implications for diagnostic testing, clinical management, and genetic counseling of patients with this devastating disorder and their families.
Figures
Comment in
-
Comment: Dravet syndrome--"old gene," novel mechanism.Neurology. 2014 Apr 8;82(14):1250. doi: 10.1212/WNL.0000000000000300. Epub 2014 Mar 12. Neurology. 2014. PMID: 24623837 Free PMC article.
-
Applications of next-generation whole exome sequencing.J Neurol. 2014 Jun;261(6):1244-6. doi: 10.1007/s00415-014-7372-1. J Neurol. 2014. PMID: 24838538 No abstract available.
References
-
- Marini C, Scheffer IE, Nabbout R, et al. The genetics of Dravet syndrome. Epilepsia 2011;52(suppl 2):24–29 - PubMed
-
- Depienne C, Gourfinkel-An I, Baulac S, LeGuern E. Genes in infantile epileptic encephalopathies. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, eds. Jasper's Basic Mechanisms of the Epilepsies. Bethesda: National Center for Biotechnology Information; 2012 - PubMed
-
- Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res 2006;70(suppl 1):S206–S217 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases