

Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability
- PMID: 24626631
- PMCID: PMC4082365
- DOI: 10.1093/hmg/ddu115
Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability
Abstract
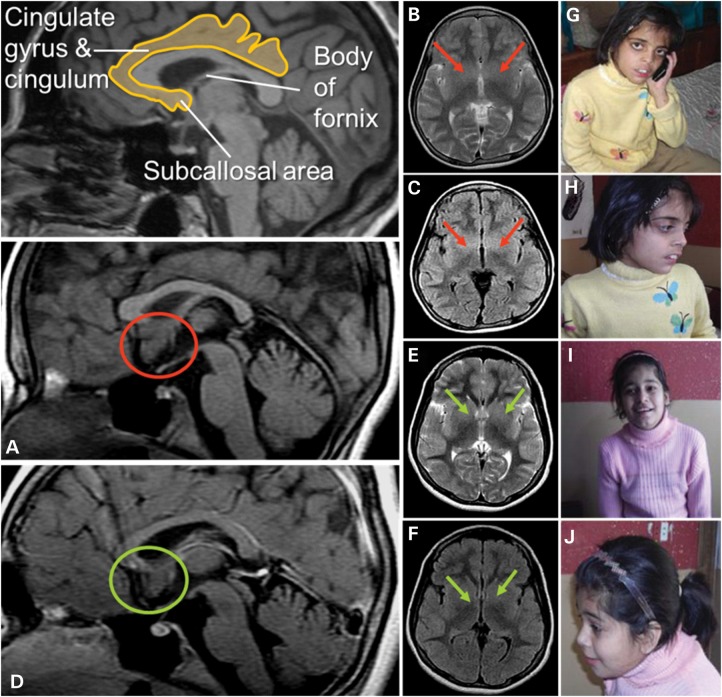
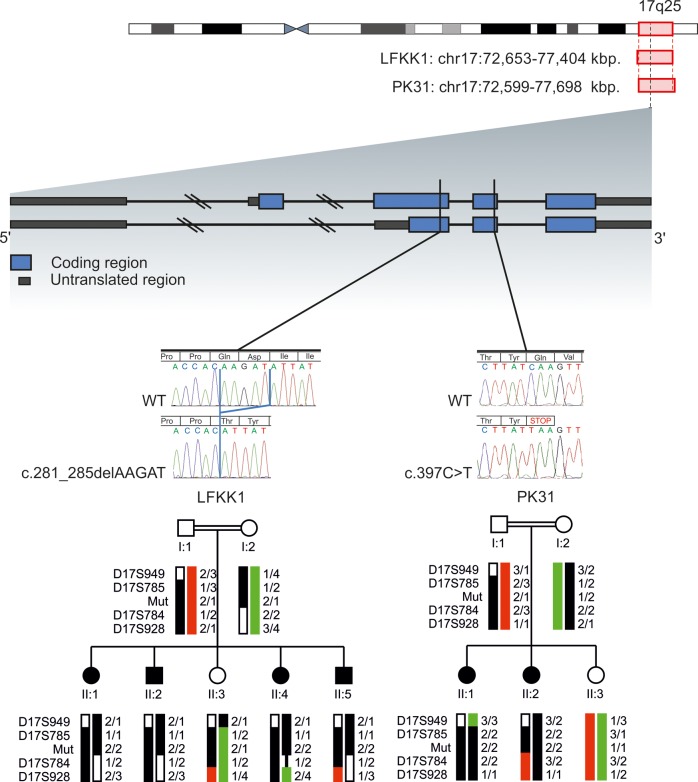
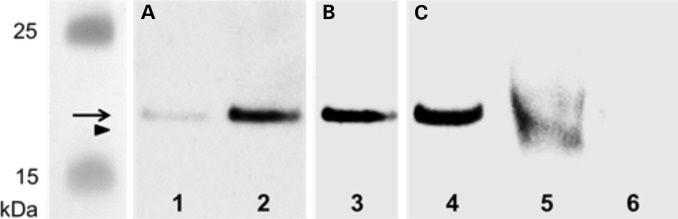
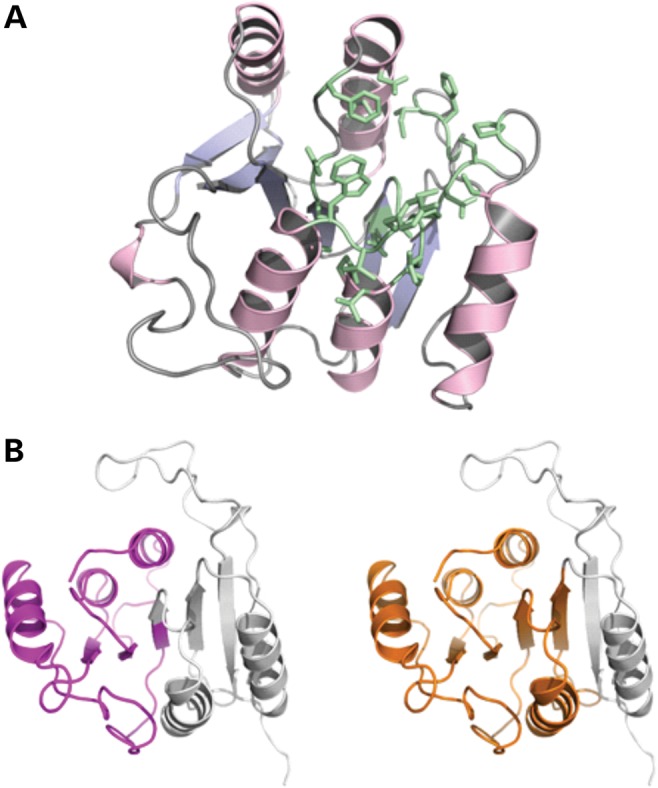
We describe the characterization of a gene for mild nonsyndromic autosomal recessive intellectual disability (ID) in two unrelated families, one from Austria, the other from Pakistan. Genome-wide single nucleotide polymorphism microarray analysis enabled us to define a region of homozygosity by descent on chromosome 17q25. Whole-exome sequencing and analysis of this region in an affected individual from the Austrian family identified a 5 bp frameshifting deletion in the METTL23 gene. By means of Sanger sequencing of METTL23, a nonsense mutation was detected in a consanguineous ID family from Pakistan for which homozygosity-by-descent mapping had identified a region on 17q25. Both changes lead to truncation of the putative METTL23 protein, which disrupts the predicted catalytic domain and alters the cellular localization. 3D-modelling of the protein indicates that METTL23 is strongly predicted to function as an S-adenosyl-methionine (SAM)-dependent methyltransferase. Expression analysis of METTL23 indicated a strong association with heat shock proteins, which suggests that these may act as a putative substrate for methylation by METTL23. A number of methyltransferases have been described recently in association with ID. Disruption of METTL23 presented here supports the importance of methylation processes for intact neuronal function and brain development.
© The Author 2014. Published by Oxford University Press.
Figures
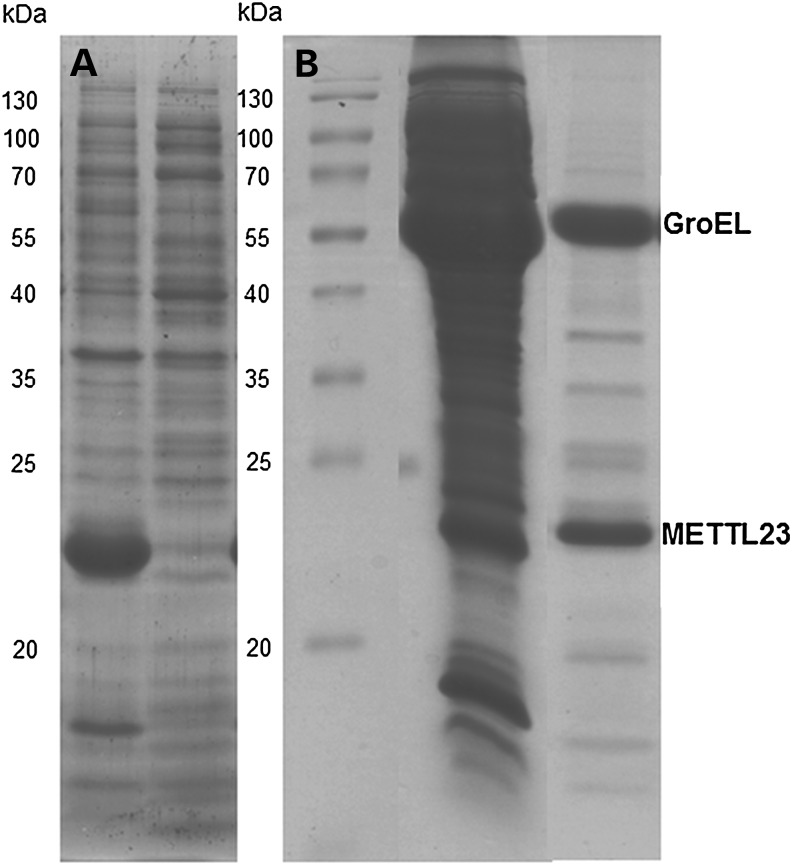
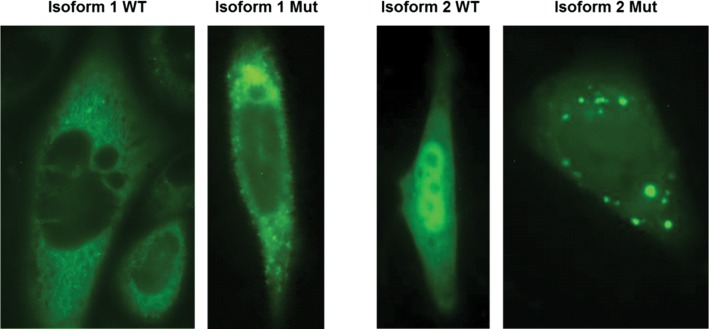
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
