

Explaining additional genetic variation in complex traits
- PMID: 24629526
- PMCID: PMC4639398
- DOI: 10.1016/j.tig.2014.02.003
Explaining additional genetic variation in complex traits
Abstract
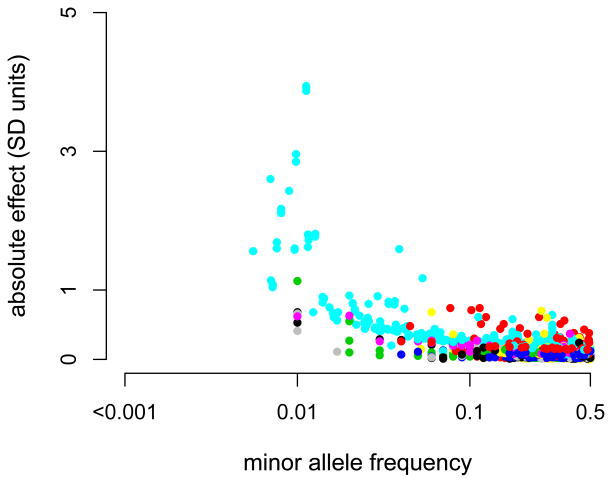
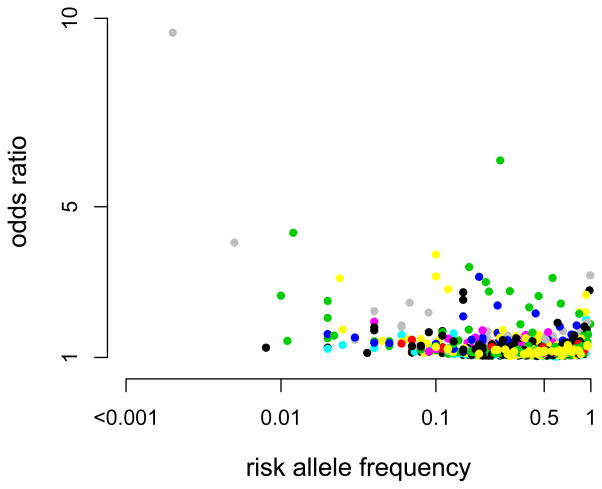
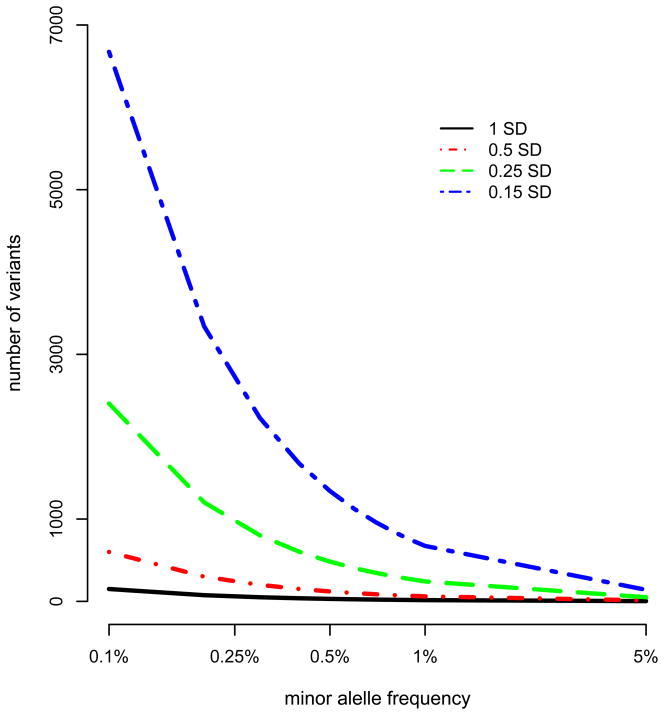
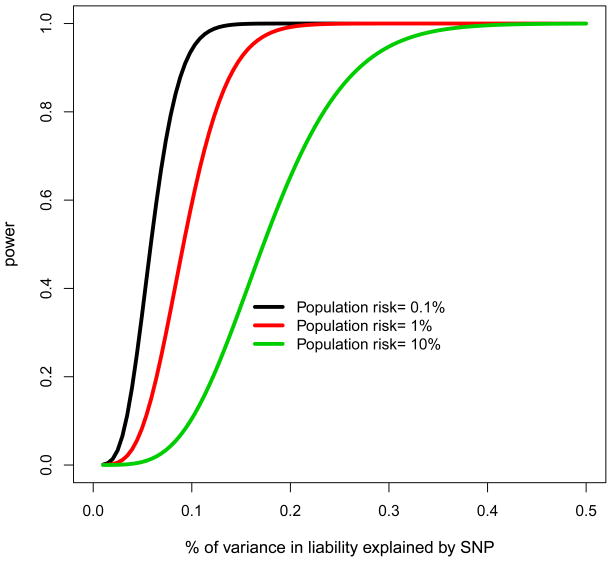
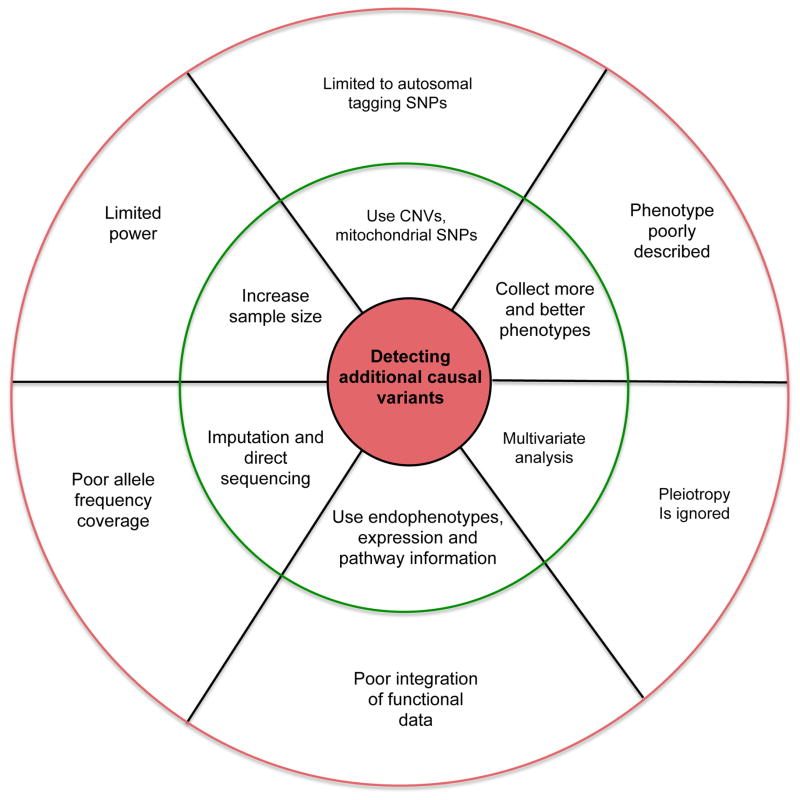
Genome-wide association studies (GWAS) have provided valuable insights into the genetic basis of complex traits, discovering >6000 variants associated with >500 quantitative traits and common complex diseases in humans. The associations identified so far represent only a fraction of those that influence phenotype, because there are likely to be many variants across the entire frequency spectrum, each of which influences multiple traits, with only a small average contribution to the phenotypic variance. This presents a considerable challenge to further dissection of the remaining unexplained genetic variance within populations, which limits our ability to predict disease risk, identify new drug targets, improve and maintain food sources, and understand natural diversity. This challenge will be met within the current framework through larger sample size, better phenotyping, including recording of nongenetic risk factors, focused study designs, and an integration of multiple sources of phenotypic and genetic information. The current evidence supports the application of quantitative genetic approaches, and we argue that one should retain simpler theories until simplicity can be traded for greater explanatory power.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
