

Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes
- PMID: 24630728
- PMCID: PMC3969382
- DOI: 10.1016/j.cell.2014.01.029
Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes
Abstract
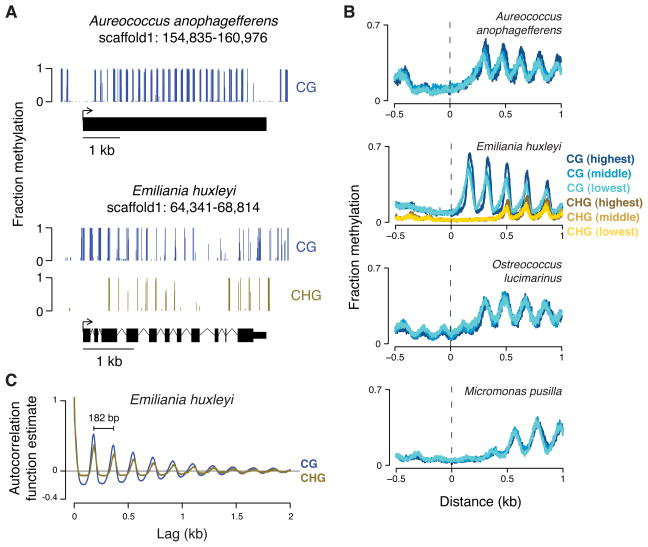
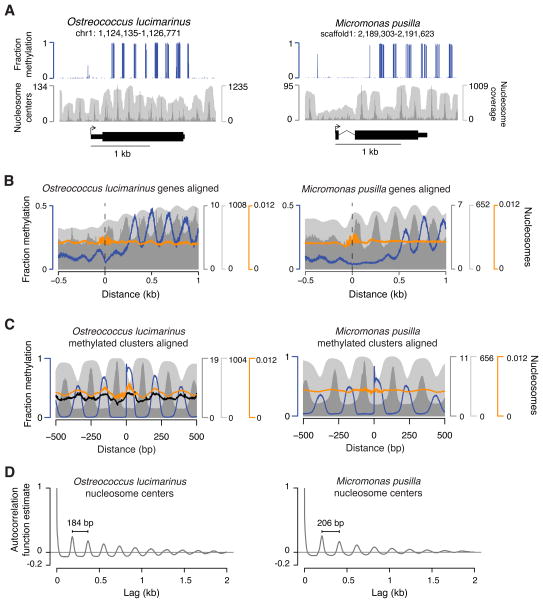
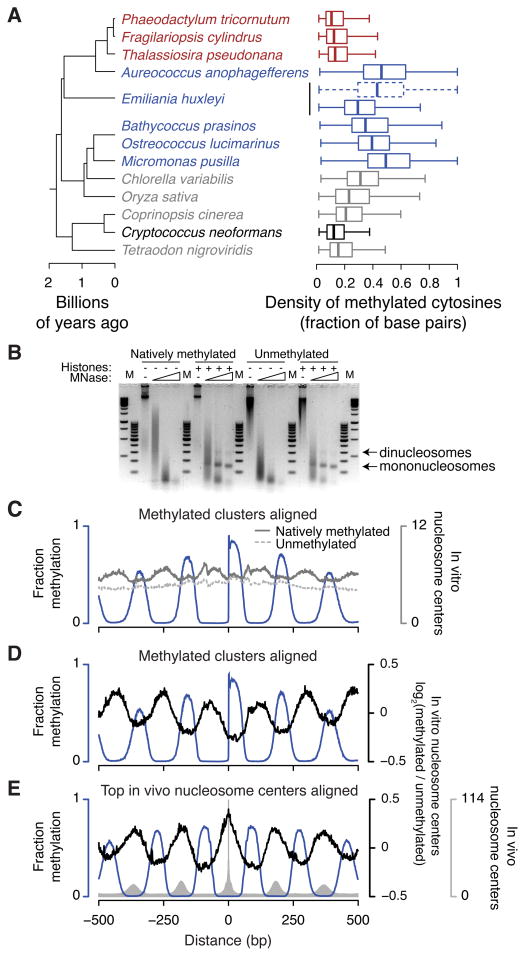
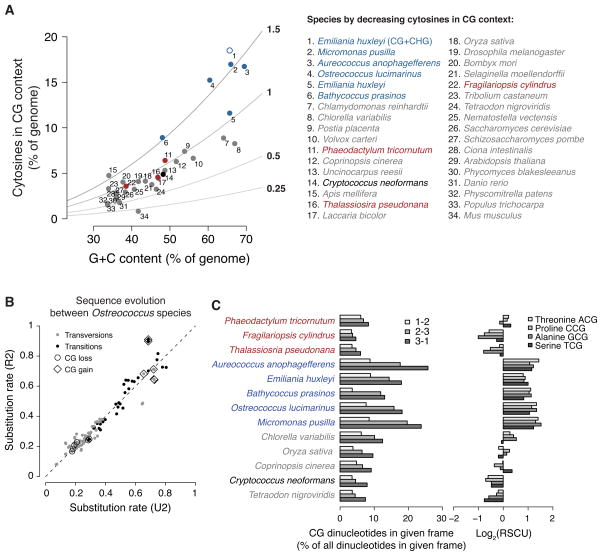
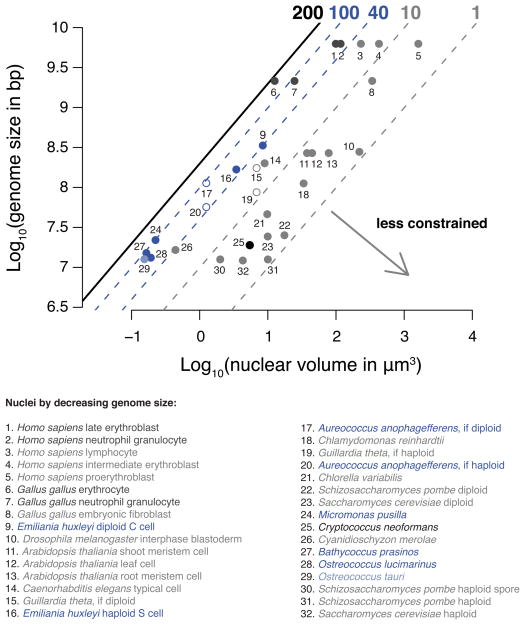
Dnmt1 epigenetically propagates symmetrical CG methylation in many eukaryotes. Their genomes are typically depleted of CG dinucleotides because of imperfect repair of deaminated methylcytosines. Here, we extensively survey diverse species lacking Dnmt1 and show that, surprisingly, symmetrical CG methylation is nonetheless frequently present and catalyzed by a different DNA methyltransferase family, Dnmt5. Numerous Dnmt5-containing organisms that diverged more than a billion years ago exhibit clustered methylation, specifically in nucleosome linkers. Clustered methylation occurs at unprecedented densities and directly disfavors nucleosomes, contributing to nucleosome positioning between clusters. Dense methylation is enabled by a regime of genomic sequence evolution that enriches CG dinucleotides and drives the highest CG frequencies known. Species with linker methylation have small, transcriptionally active nuclei that approach the physical limits of chromatin compaction. These features constitute a previously unappreciated genome architecture, in which dense methylation influences nucleosome positions, likely facilitating nuclear processes under extreme spatial constraints.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures


References
-
- Charlesworth B, Charlesworth D. Elements of Evolutionary Genetics. Roberts and Company Publishers; 2010.
-
- Cohen NM, Kenigsberg E, Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145:773–786. - PubMed
-
- Coleman-Derr D, Zilberman D. DNA methylation, H2A.Z, and the regulation of constitutive expression. Cold Spring Harb Symp Quant Biol 2012 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
