

Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs
- PMID: 24631770
- PMCID: PMC4080911
- DOI: 10.1016/j.yjmcc.2014.02.015
Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs
Abstract
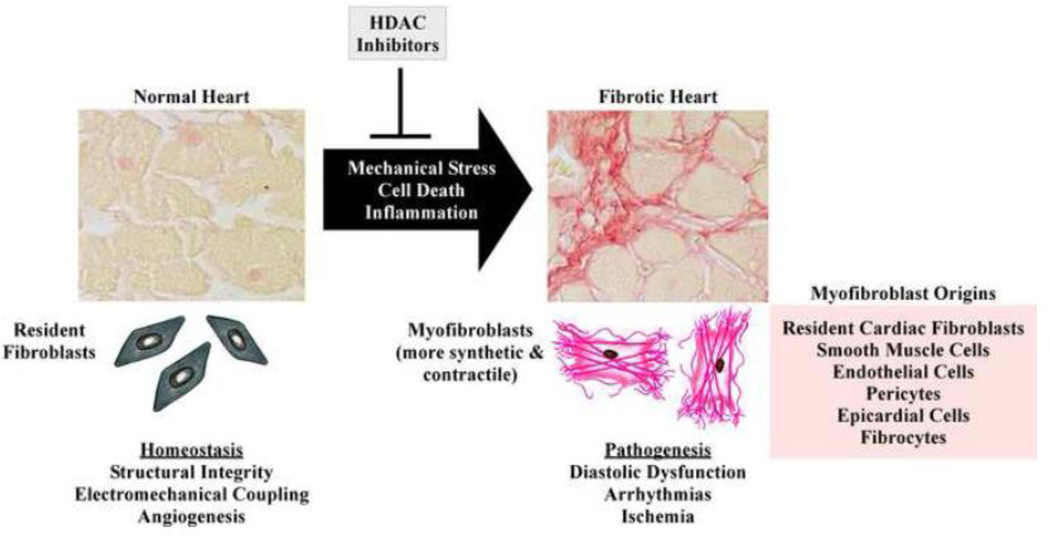
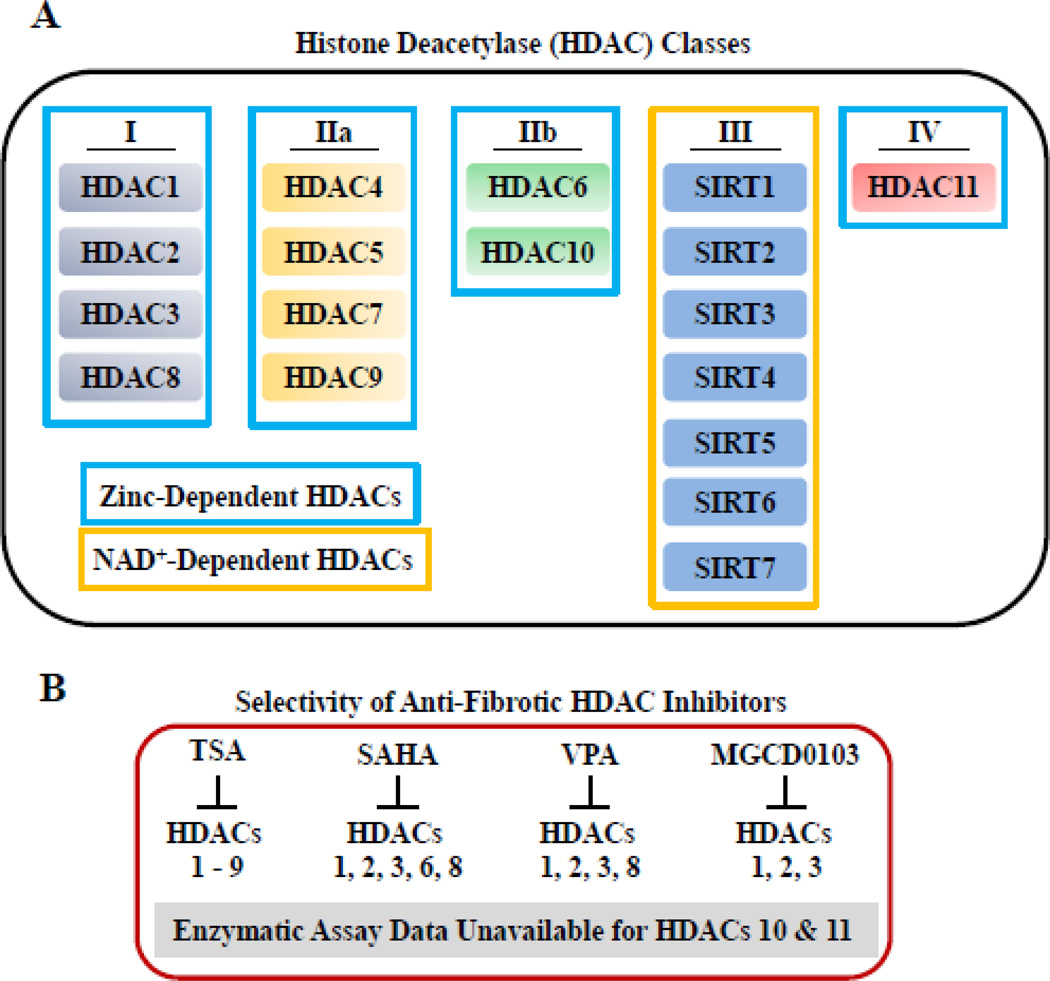
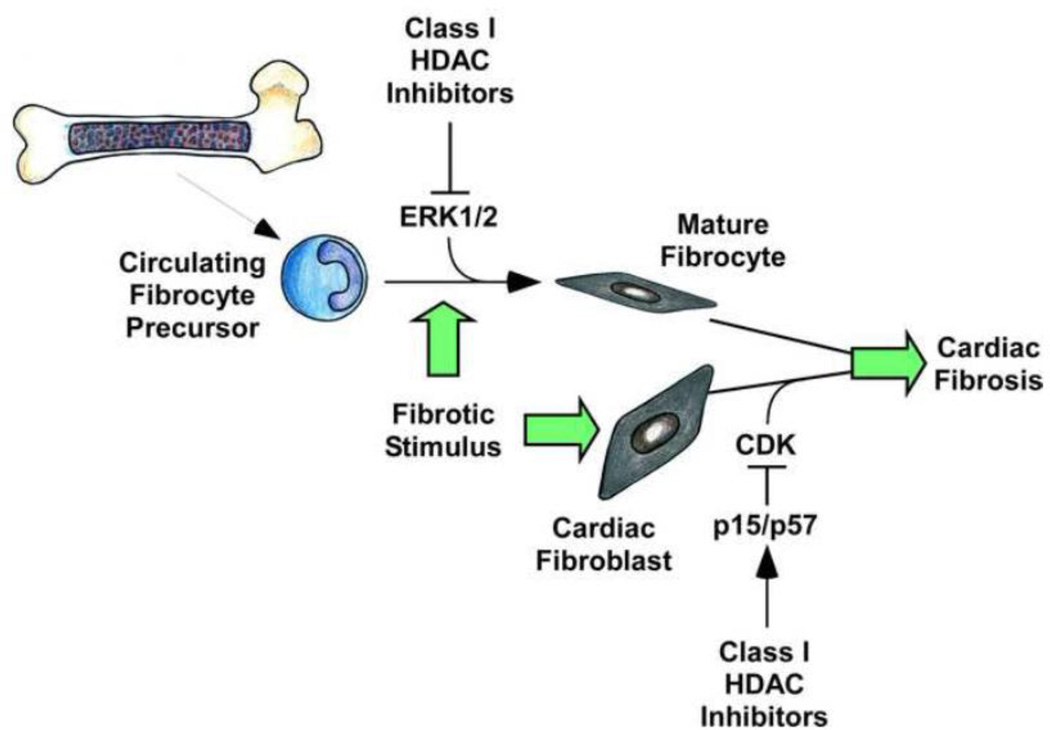
Cardiac fibrosis is implicated in numerous physiologic and pathologic conditions, including scar formation, heart failure and cardiac arrhythmias. However the specific cells and signaling pathways mediating this process are poorly understood. Lysine acetylation of nucleosomal histone tails is an important mechanism for the regulation of gene expression. Additionally, proteomic studies have revealed that thousands of proteins in all cellular compartments are subject to reversible lysine acetylation, and thus it is becoming clear that this post-translational modification will rival phosphorylation in terms of biological import. Acetyl groups are conjugated to lysine by histone acetyltransferases (HATs) and removed from lysine by histone deacetylases (HDACs). Recent studies have shown that pharmacologic agents that alter lysine acetylation by targeting HDACs have the remarkable ability to block pathological fibrosis. Here, we review the current understanding of cardiac fibroblasts and the fibrogenic process with respect to the roles of lysine acetylation in the control of disease-related cardiac fibrosis. Potential for small molecule HDAC inhibitors as anti-fibrotic therapeutics that target cardiac fibroblasts is highlighted. This article is part of a Special Issue entitled "Myocyte-Fibroblast Signalling in Myocardium."
Keywords: Epigenetics; Fibroblast; Fibrosis; Histone acetylation.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Conflict of interest statement
No conflicts of interest exist for the authors.
Figures
References
-
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–842. - PubMed
-
- Maass AH, Leinwand LA. Mechanisms of the pathogenesis of troponin T-based familial hypertrophic cardiomyopathy. Trends Cardiovasc Med. 2003;13:232–237. - PubMed
-
- Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. - PubMed
-
- Virchow R. Die Cellularpathologie in ihrer Begrundung auf physiologische und pathologische Gewebelehre. Berlin. 1858 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
