

The many faces of autophagy dysfunction in Huntington's disease: from mechanism to therapy
- PMID: 24632005
- PMCID: PMC4096219
- DOI: 10.1016/j.drudis.2014.02.014
The many faces of autophagy dysfunction in Huntington's disease: from mechanism to therapy
Abstract
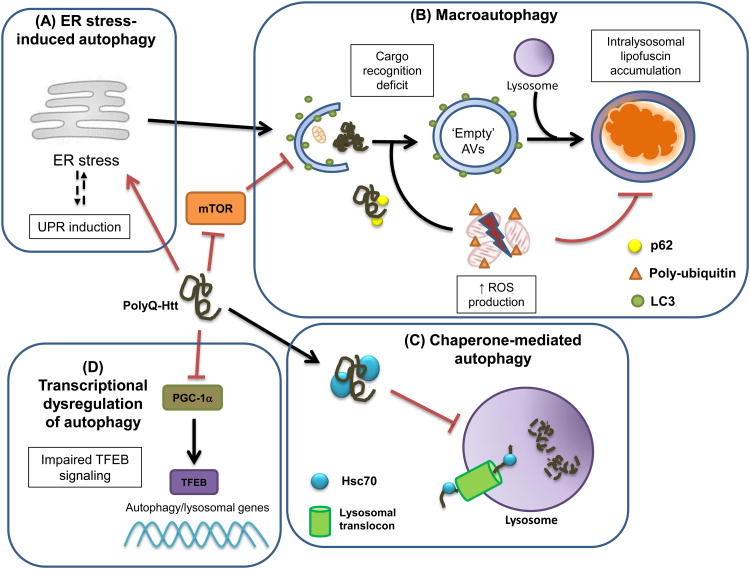
Autophagy is the cellular process by which proteins, macromolecules, and organelles are targeted to and degraded by the lysosome. Given that neurodegenerative diseases involve the production of misfolded proteins that cannot be degraded by the protein quality-control systems of the cell, the autophagy pathway is now the focus of intense scrutiny, because autophagy is primarily responsible for maintaining normal cellular proteostasis in the central nervous system (CNS). Huntington's disease (HD) is an inherited CAG-polyglutamine repeat disorder, resulting from the production and accumulation of misfolded huntingtin (Htt) protein. HD shares key features with common neurodegenerative disorders, such as Alzheimer's disease (AD) and Parkinson's disease (PD) and, thus, belongs to a large class of disorders known as neurodegenerative proteinopathies. Multiple independent lines of research have documented alterations in autophagy function in HD, and numerous studies have demonstrated a potential role for autophagy modulation as a therapeutic intervention. In this review, we consider the evidence for autophagy dysfunction in HD, and delineate different targets and mechanistic pathways that might account for the autophagy abnormalities detected in HD. We assess the utility of autophagy modulation as a treatment modality in HD, and suggest guidelines and caveats for future therapy development directed at the autophagy pathway in HD and related disorders.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Degradation of misfolded proteins by autophagy: is it a strategy for Huntington's disease treatment?J Huntingtons Dis. 2013;2(2):149-57. doi: 10.3233/JHD-130052. J Huntingtons Dis. 2013. PMID: 25063512 Review.
-
Endoplasmic reticulum stress: The cause and solution to Huntington's disease?Brain Res. 2016 Oct 1;1648(Pt B):650-657. doi: 10.1016/j.brainres.2016.03.034. Epub 2016 Apr 1. Brain Res. 2016. PMID: 27040914 Review.
-
Autophagy in Huntington disease and huntingtin in autophagy.Trends Neurosci. 2015 Jan;38(1):26-35. doi: 10.1016/j.tins.2014.09.003. Epub 2014 Oct 2. Trends Neurosci. 2015. PMID: 25282404 Review.
-
Proteostasis in Huntington's disease: disease mechanisms and therapeutic opportunities.Acta Pharmacol Sin. 2018 May;39(5):754-769. doi: 10.1038/aps.2018.11. Epub 2018 Apr 5. Acta Pharmacol Sin. 2018. PMID: 29620053 Free PMC article. Review.
-
Targeting the proteostasis network in Huntington's disease.Ageing Res Rev. 2019 Jan;49:92-103. doi: 10.1016/j.arr.2018.11.006. Epub 2018 Nov 28. Ageing Res Rev. 2019. PMID: 30502498 Free PMC article. Review.
Cited by
-
Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions.Biomedicines. 2021 Nov 5;9(11):1625. doi: 10.3390/biomedicines9111625. Biomedicines. 2021. PMID: 34829854 Free PMC article. Review.
-
The Regulation of MiTF/TFE Transcription Factors Across Model Organisms: from Brain Physiology to Implication for Neurodegeneration.Mol Neurobiol. 2022 Aug;59(8):5000-5023. doi: 10.1007/s12035-022-02895-3. Epub 2022 Jun 4. Mol Neurobiol. 2022. PMID: 35665902 Free PMC article. Review.
-
Biological Aging and the Cellular Pathogenesis of Huntington's Disease.J Huntingtons Dis. 2020;9(2):115-128. doi: 10.3233/JHD-200395. J Huntingtons Dis. 2020. PMID: 32417788 Free PMC article. Review.
-
Lysosomal Biology and Function: Modern View of Cellular Debris Bin.Cells. 2020 May 4;9(5):1131. doi: 10.3390/cells9051131. Cells. 2020. PMID: 32375321 Free PMC article. Review.
-
MALAT1 Mediates α-Synuclein Expression through miR-23b-3p to Induce Autophagic Impairment and the Inflammatory Response in Microglia to Promote Apoptosis in Dopaminergic Neuronal Cells.Mediators Inflamm. 2023 Apr 6;2023:4477492. doi: 10.1155/2023/4477492. eCollection 2023. Mediators Inflamm. 2023. PMID: 37064502 Free PMC article.
References
-
- Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. - PubMed
-
- Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. - PubMed
-
- Sapp E, et al. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42:604–612. - PubMed
-
- DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
