

Genome-guided transcript assembly by integrative analysis of RNA sequence data
- PMID: 24633242
- PMCID: PMC4037530
- DOI: 10.1038/nbt.2850
Genome-guided transcript assembly by integrative analysis of RNA sequence data
Abstract
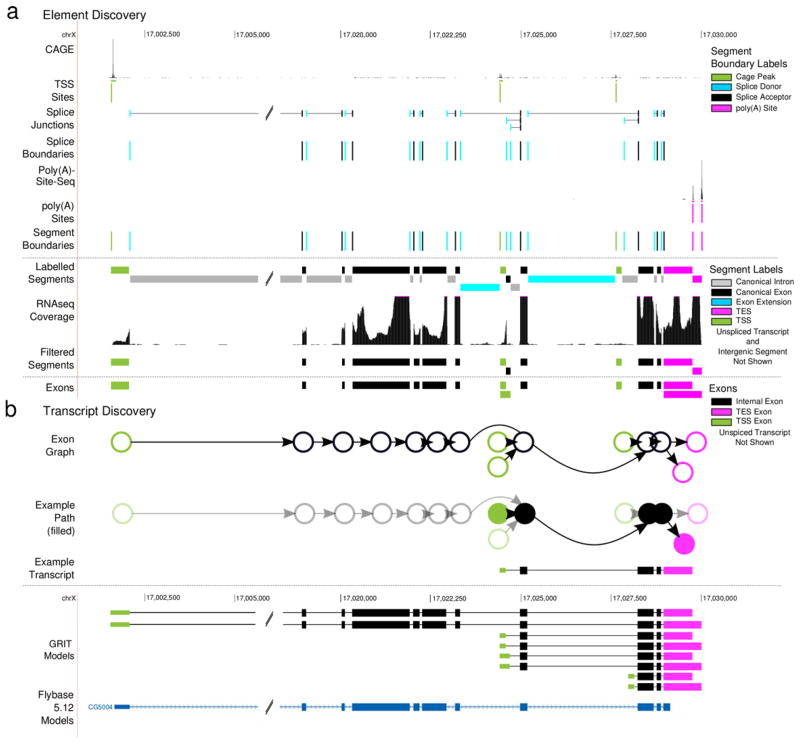
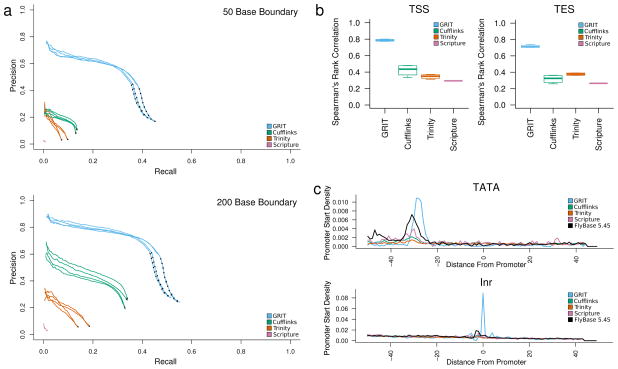
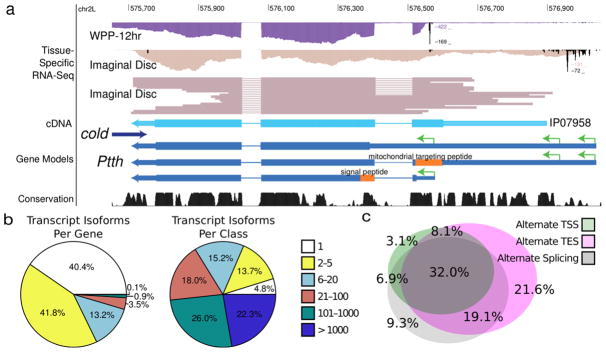
The identification of full length transcripts entirely from short-read RNA sequencing data (RNA-seq) remains a challenge in the annotation of genomes. Here we describe an automated pipeline for genome annotation that integrates RNA-seq and gene-boundary data sets, which we call Generalized RNA Integration Tool, or GRIT. Applying GRIT to Drosophila melanogaster short-read RNA-seq, cap analysis of gene expression (CAGE) and poly(A)-site-seq data collected for the modENCODE project, we recovered the vast majority of previously annotated transcripts and doubled the total number of transcripts cataloged. We found that 20% of protein coding genes encode multiple protein-localization signals and that, in 20-d-old adult fly heads, genes with multiple polyadenylation sites are more common than genes with alternative splicing or alternative promoters. GRIT demonstrates 30% higher precision and recall than the most widely used transcript assembly tools. GRIT will facilitate the automated generation of high-quality genome annotations without the need for extensive manual annotation.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5 (7):621–628. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
