

RepARK--de novo creation of repeat libraries from whole-genome NGS reads
- PMID: 24634442
- PMCID: PMC4027187
- DOI: 10.1093/nar/gku210
RepARK--de novo creation of repeat libraries from whole-genome NGS reads
Abstract
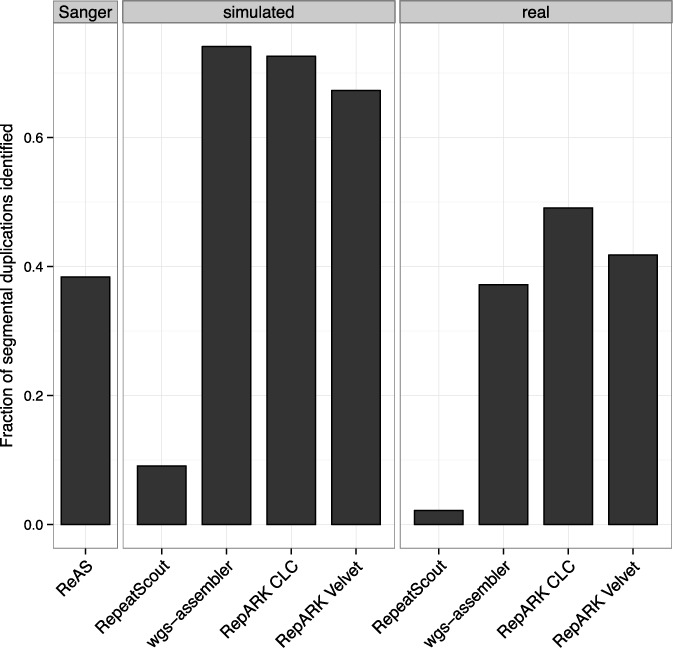
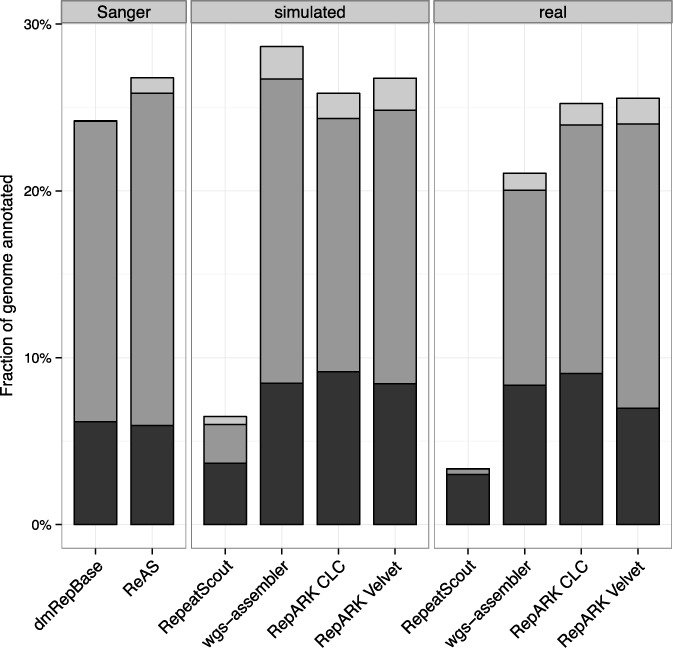
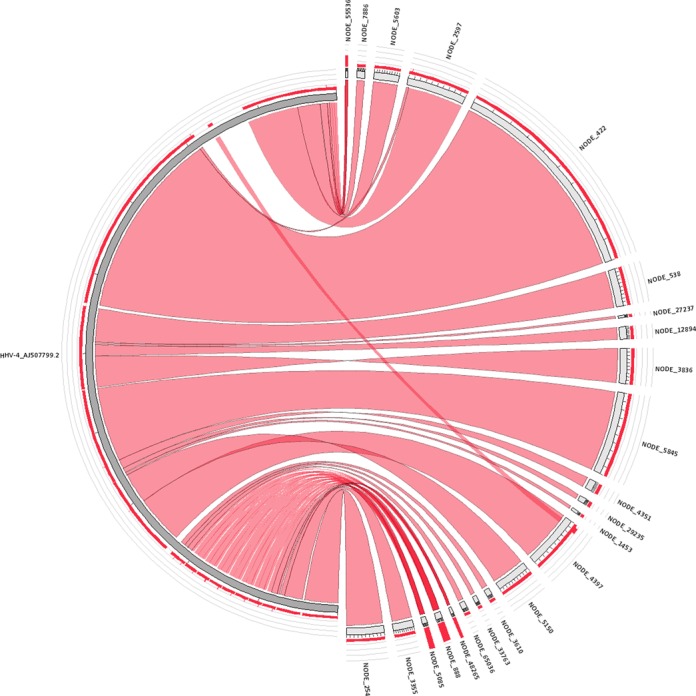
Generation of repeat libraries is a critical step for analysis of complex genomes. In the era of next-generation sequencing (NGS), such libraries are usually produced using a whole-genome shotgun (WGS) derived reference sequence whose completeness greatly influences the quality of derived repeat libraries. We describe here a de novo repeat assembly method--RepARK (Repetitive motif detection by Assembly of Repetitive K-mers)--which avoids potential biases by using abundant k-mers of NGS WGS reads without requiring a reference genome. For validation, repeat consensuses derived from simulated and real Drosophila melanogaster NGS WGS reads were compared to repeat libraries generated by four established methods. RepARK is orders of magnitude faster than the other methods and generates libraries that are: (i) composed almost entirely of repetitive motifs, (ii) more comprehensive and (iii) almost completely annotated by TEclass. Additionally, we show that the RepARK method is applicable to complex genomes like human and can even serve as a diagnostic tool to identify repetitive sequences contaminating NGS datasets.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
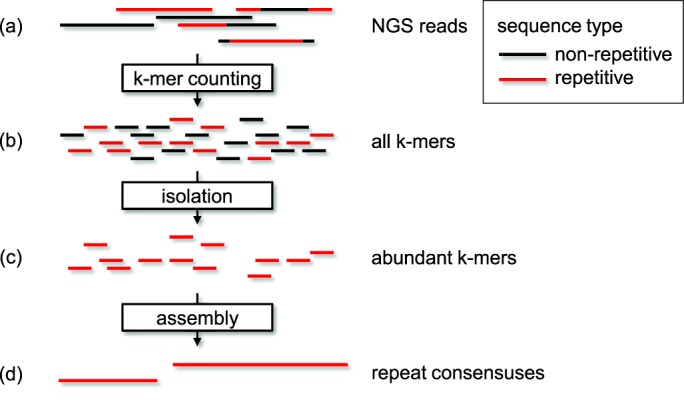
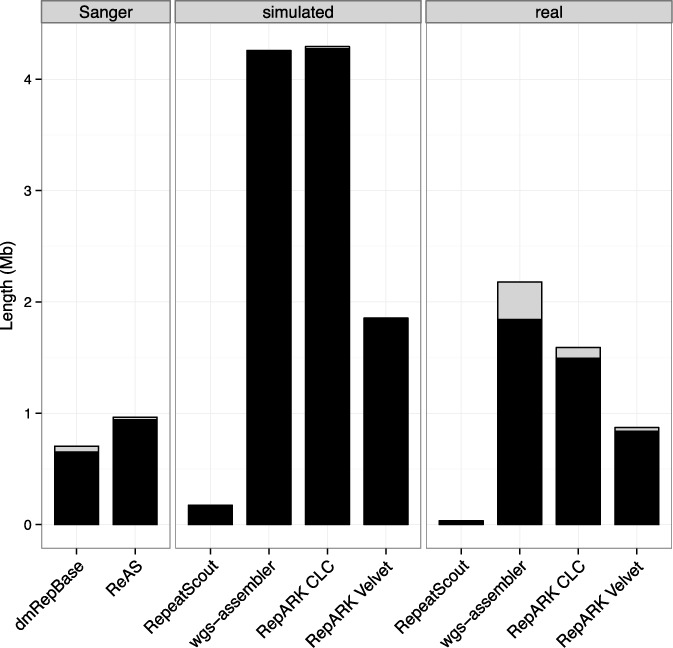
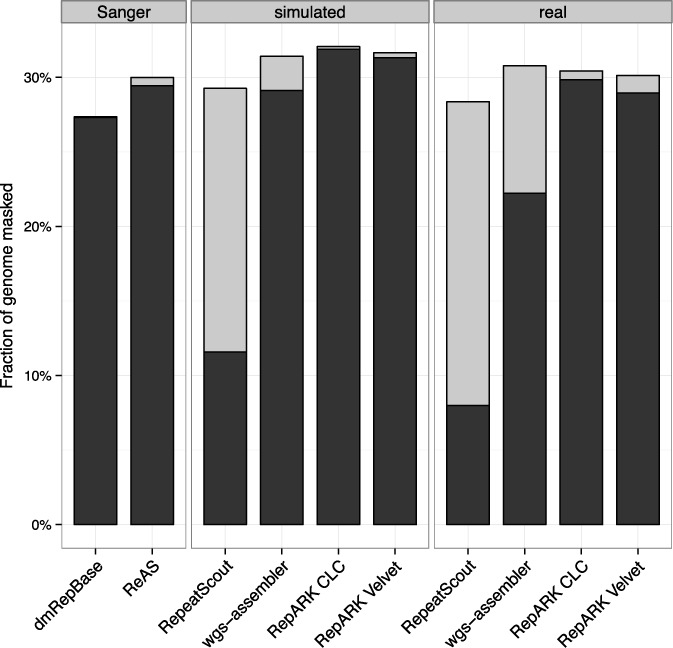
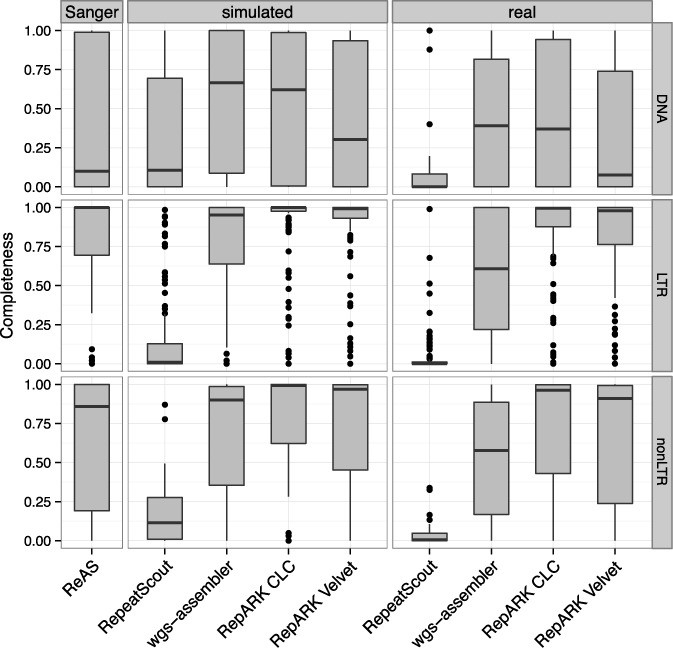
References
-
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Mayer K.F., Waugh R., Brown J.W., Schulman A., Langridge P., Platzer M., Fincher G.B., Muehlbauer G.J., Sato K., Close T.J., et al. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–716. - PubMed
-
- Yandell M., Ence D. A beginner's guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012;13:329–342. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
