

De novo structure prediction of globular proteins aided by sequence variation-derived contacts
- PMID: 24637808
- PMCID: PMC3956894
- DOI: 10.1371/journal.pone.0092197
De novo structure prediction of globular proteins aided by sequence variation-derived contacts
Abstract
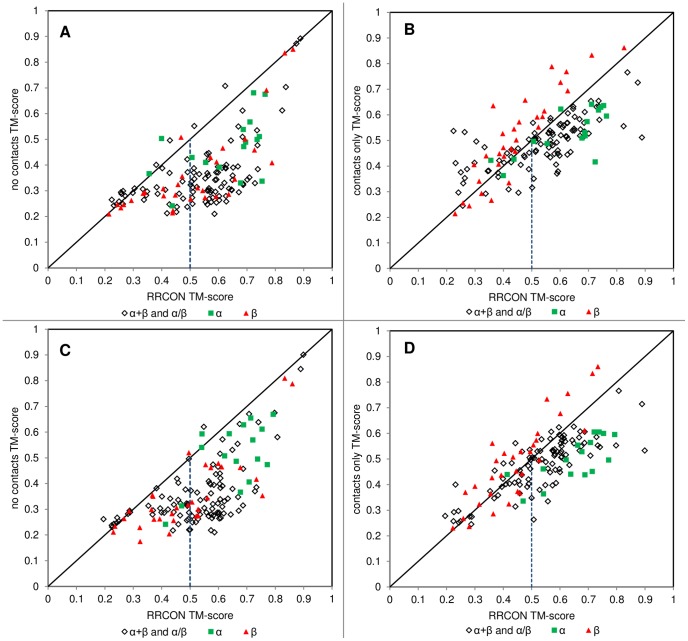
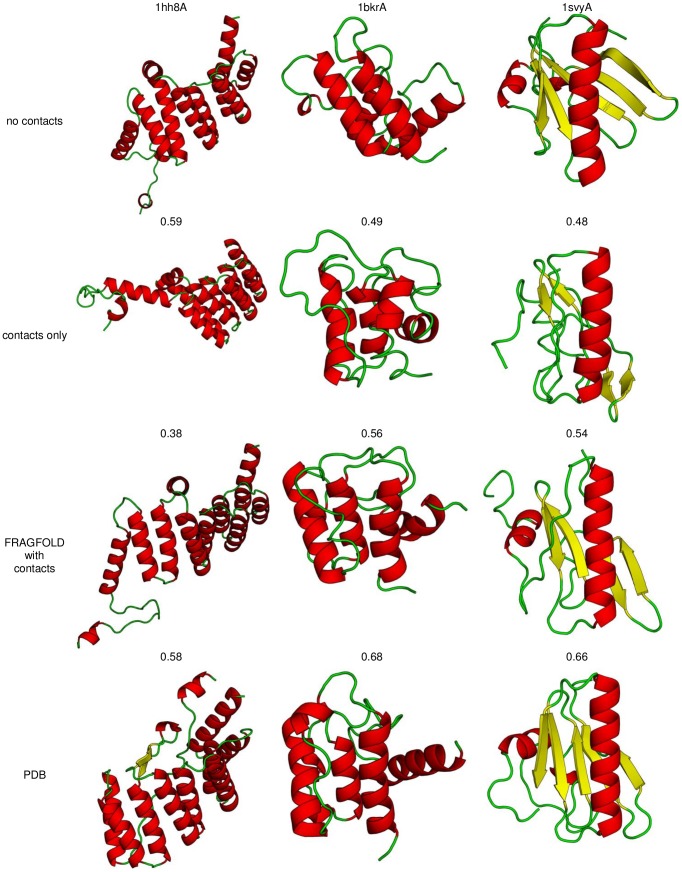
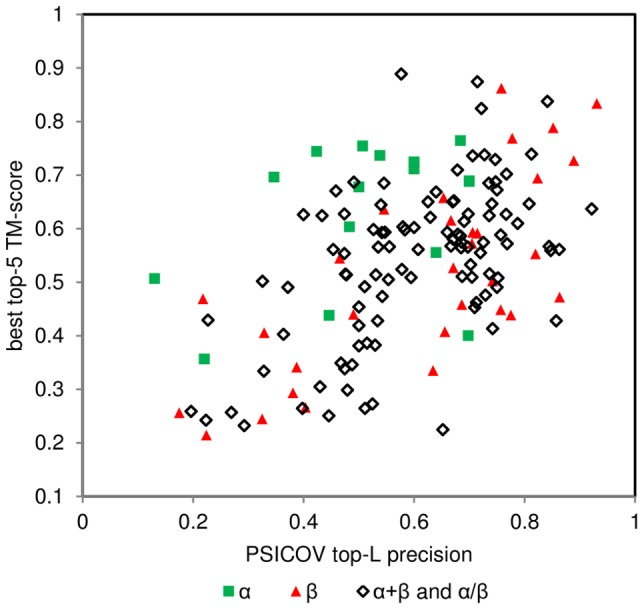
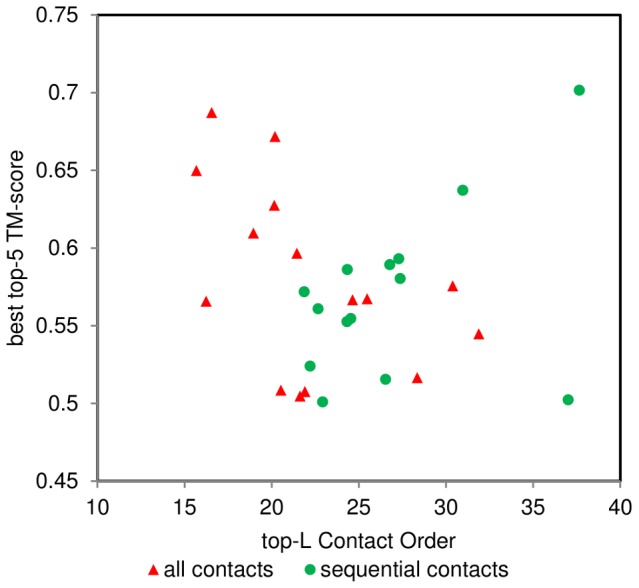
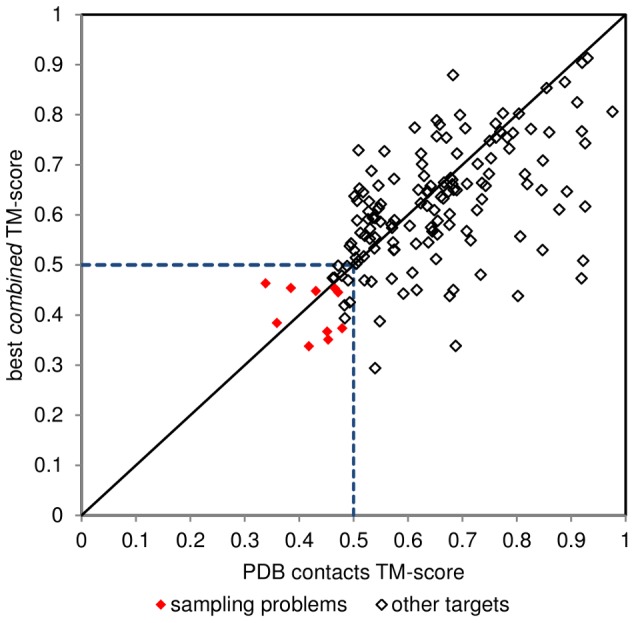
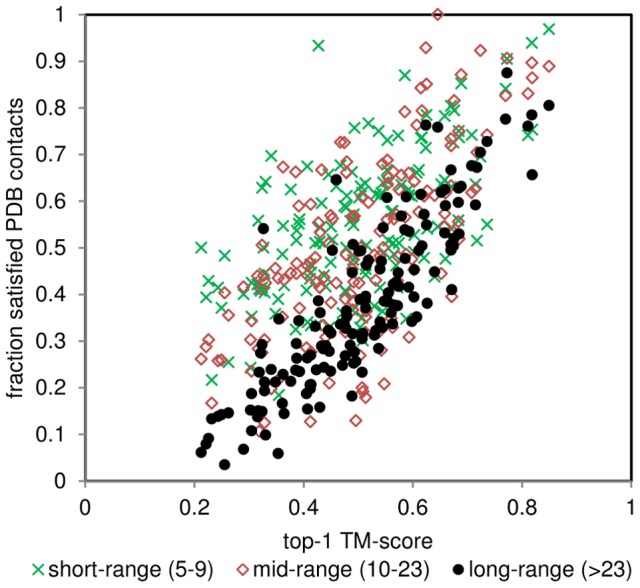
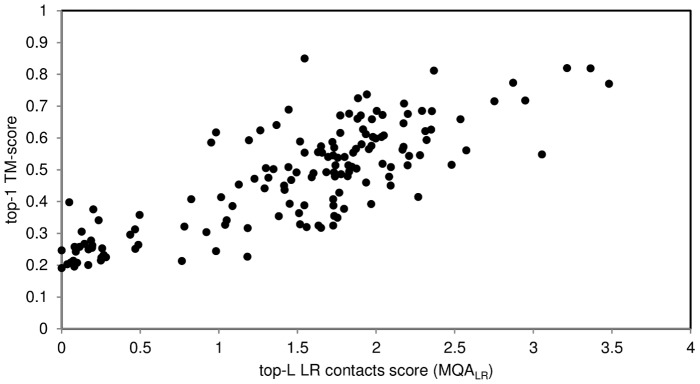
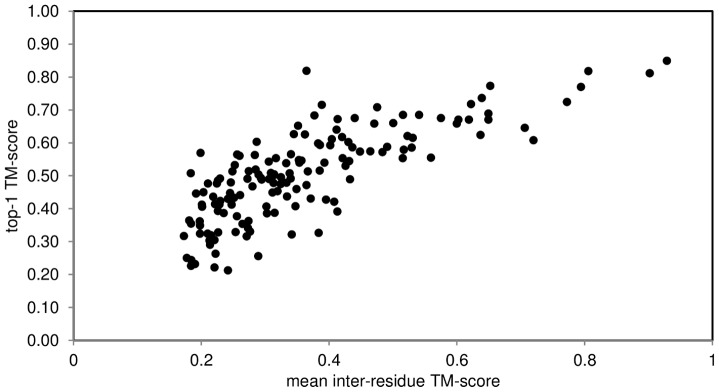
The advent of high accuracy residue-residue intra-protein contact prediction methods enabled a significant boost in the quality of de novo structure predictions. Here, we investigate the potential benefits of combining a well-established fragment-based folding algorithm--FRAGFOLD, with PSICOV, a contact prediction method which uses sparse inverse covariance estimation to identify co-varying sites in multiple sequence alignments. Using a comprehensive set of 150 diverse globular target proteins, up to 266 amino acids in length, we are able to address the effectiveness and some limitations of such approaches to globular proteins in practice. Overall we find that using fragment assembly with both statistical potentials and predicted contacts is significantly better than either statistical potentials or contacts alone. Results show up to nearly 80% of correct predictions (TM-score ≥0.5) within analysed dataset and a mean TM-score of 0.54. Unsuccessful modelling cases emerged either from conformational sampling problems, or insufficient contact prediction accuracy. Nevertheless, a strong dependency of the quality of final models on the fraction of satisfied predicted long-range contacts was observed. This not only highlights the importance of these contacts on determining the protein fold, but also (combined with other ensemble-derived qualities) provides a powerful guide as to the choice of correct models and the global quality of the selected model. A proposed quality assessment scoring function achieves 0.93 precision and 0.77 recall for the discrimination of correct folds on our dataset of decoys. These findings suggest the approach is well-suited for blind predictions on a variety of globular proteins of unknown 3D structure, provided that enough homologous sequences are available to construct a large and accurate multiple sequence alignment for the initial contact prediction step.
Conflict of interest statement
Figures


References
-
- Göbel U, Sander C, Schneider R, Valencia A (1994) Correlated mutations and residue contacts in proteins. Proteins 18: 309–317. - PubMed
-
- Pollock DD, Taylor WR (1997) Effectiveness of correlation analysis in identifying protein residues undergoing correlated evolution. Protein Eng 10: 647–657. - PubMed
-
- Gromiha MM, Selvaraj S (2004) Inter-residue interactions in protein folding and stability. Prog Biophys Mol Biol 86: 235–277. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
