

TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia
- PMID: 24637876
- PMCID: PMC3956758
- DOI: 10.1371/journal.pone.0091598
TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia
Abstract
Background: Although TBX1 mutations have been identified in patients with 22q11.2 deletion syndrome (22q11.2DS)-like phenotypes including characteristic craniofacial features, cardiovascular anomalies, hypoparathyroidism, and thymic hypoplasia, the frequency of TBX1 mutations remains rare in deletion-negative patients. Thus, it would be reasonable to perform a comprehensive genetic analysis in deletion-negative patients with 22q11.2DS-like phenotypes.
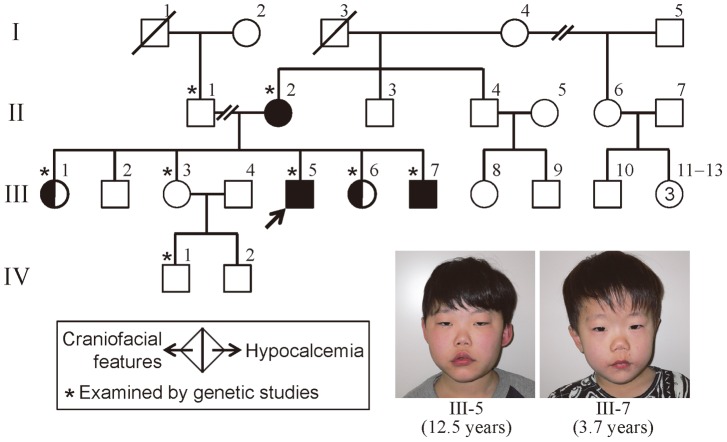
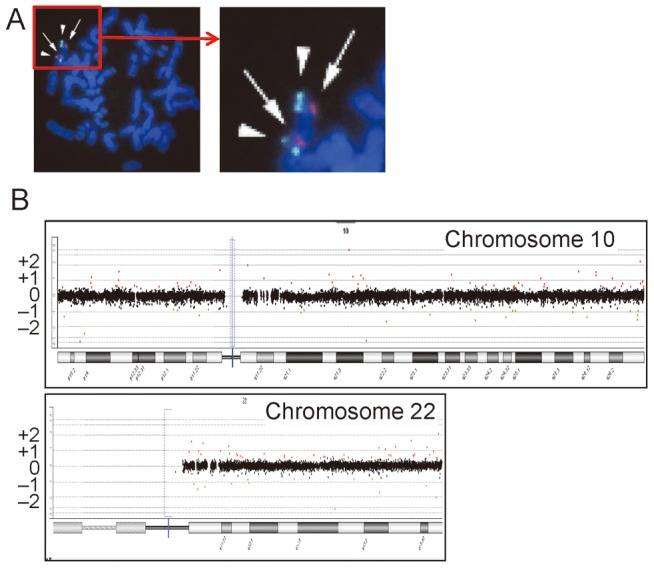
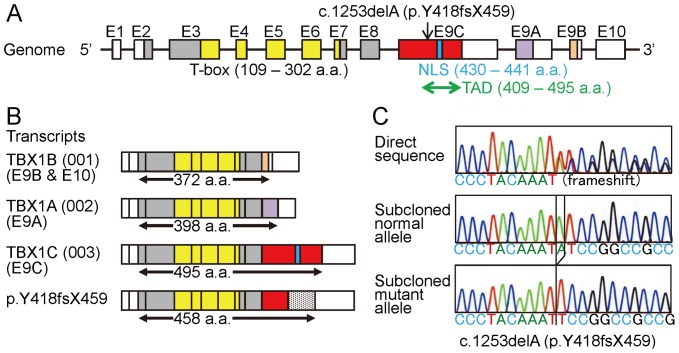
Methodology/principal findings: We studied three subjects with craniofacial features and hypocalcemia (group 1), two subjects with craniofacial features alone (group 2), and three subjects with normal phenotype within a single Japanese family. Fluorescence in situ hybridization analysis excluded chromosome 22q11.2 deletion, and genomewide array comparative genomic hybridization analysis revealed no copy number change specific to group 1 or groups 1+2. However, exome sequencing identified a heterozygous TBX1 frameshift mutation (c.1253delA, p.Y418fsX459) specific to groups 1+2, as well as six missense variants and two in-frame microdeletions specific to groups 1+2 and two missense variants specific to group 1. The TBX1 mutation resided at exon 9C and was predicted to produce a non-functional truncated protein missing the nuclear localization signal and most of the transactivation domain.
Conclusions/significance: Clinical features in groups 1+2 are well explained by the TBX1 mutation, while the clinical effects of the remaining variants are largely unknown. Thus, the results exemplify the usefulness of exome sequencing in the identification of disease-causing mutations in familial disorders. Furthermore, the results, in conjunction with the previous data, imply that TBX1 isoform C is the biologically essential variant and that TBX1 mutations are associated with a wide phenotypic spectrum, including most of 22q11.2DS phenotypes.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
