

Aβ-induced Golgi fragmentation in Alzheimer's disease enhances Aβ production
- PMID: 24639524
- PMCID: PMC3977293
- DOI: 10.1073/pnas.1320192111
Aβ-induced Golgi fragmentation in Alzheimer's disease enhances Aβ production
Abstract
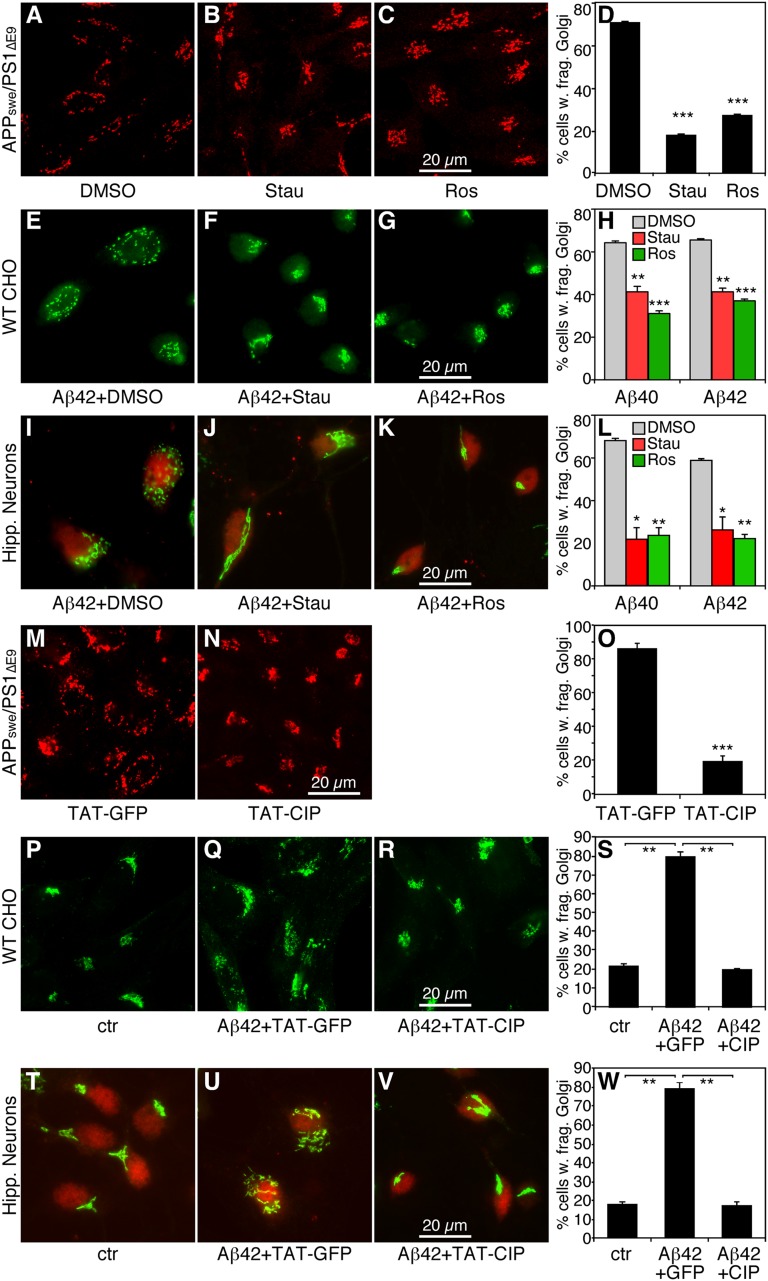
Golgi fragmentation occurs in neurons of patients with Alzheimer's disease (AD), but the underlying molecular mechanism causing the defects and the subsequent effects on disease development remain unknown. In this study, we examined the Golgi structure in APPswe/PS1E9 transgenic mouse and tissue culture models. Our results show that accumulation of amyloid beta peptides (Aβ) leads to Golgi fragmentation. Further biochemistry and cell biology studies revealed that Golgi fragmentation in AD is caused by phosphorylation of Golgi structural proteins, such as GRASP65, which is induced by Aβ-triggered cyclin-dependent kinase-5 activation. Significantly, both inhibition of cyclin-dependent kinase-5 and expression of nonphosphorylatable GRASP65 mutants rescued the Golgi structure and reduced Aβ secretion by elevating α-cleavage of the amyloid precursor protein. Our study demonstrates a molecular mechanism for Golgi fragmentation and its effects on amyloid precursor protein trafficking and processing in AD, suggesting Golgi as a potential drug target for AD treatment.
Keywords: APP processing; GRASP55; Golgi stacking; amyloidogenic.
Conflict of interest statement
The authors declare no conflict of interest.
Figures







References
-
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells: Evidence that cleavage at the “beta-secretase” site occurs in the Golgi apparatus. J Biol Chem. 1996;271(16):9390–9397. - PubMed
-
- Schmidt V, et al. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem. 2007;282(45):32956–32964. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
