

Design and synthesis of high affinity inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferases directed by ligand efficiency dependent lipophilicity (LELP)
- PMID: 24641010
- PMCID: PMC4048319
- DOI: 10.1021/jm500066b
Design and synthesis of high affinity inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferases directed by ligand efficiency dependent lipophilicity (LELP)
Abstract
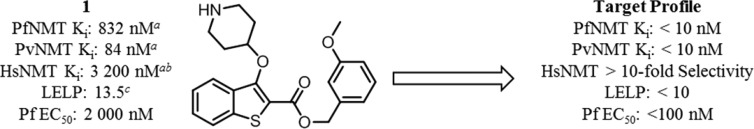
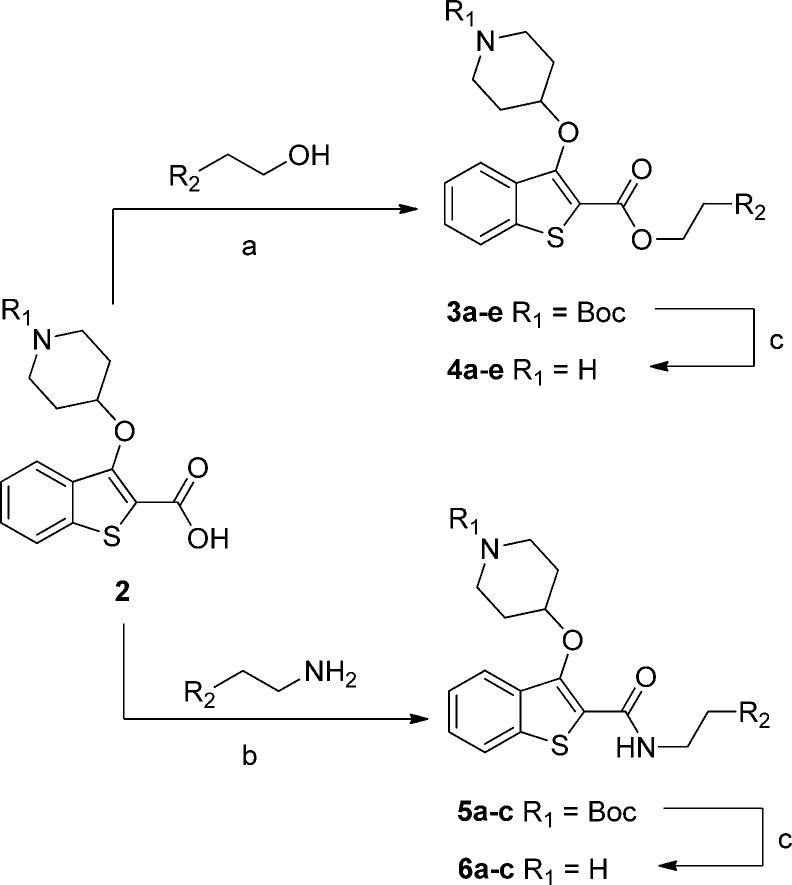
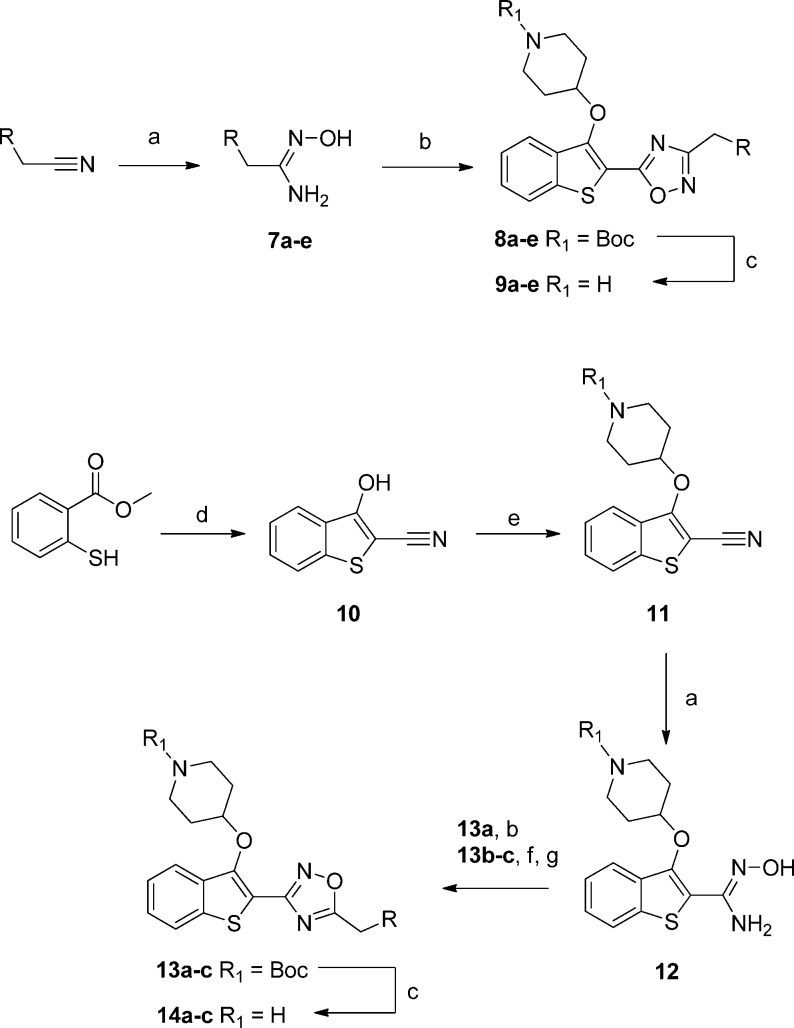
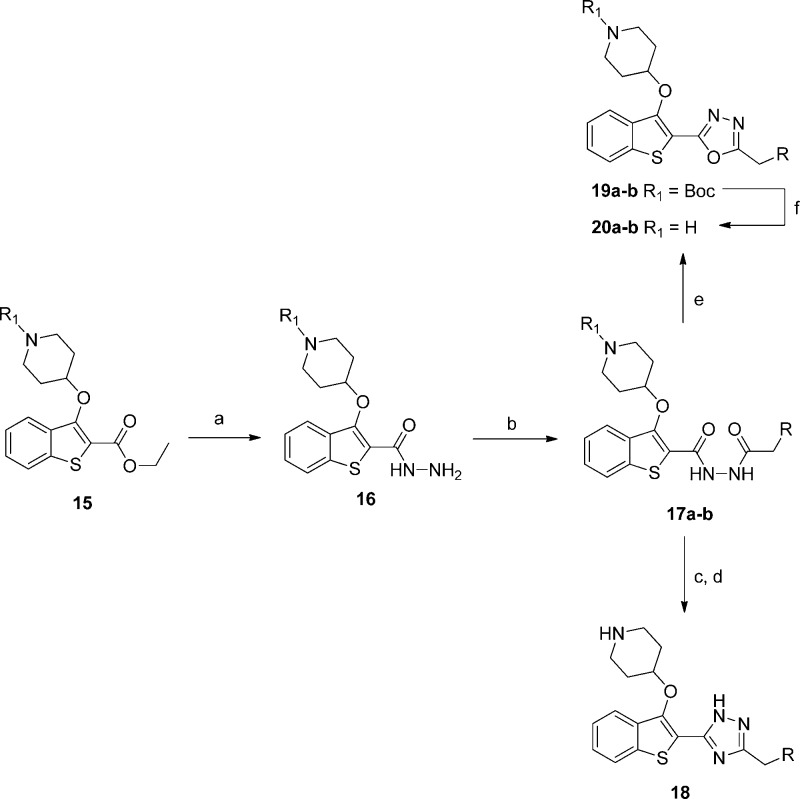
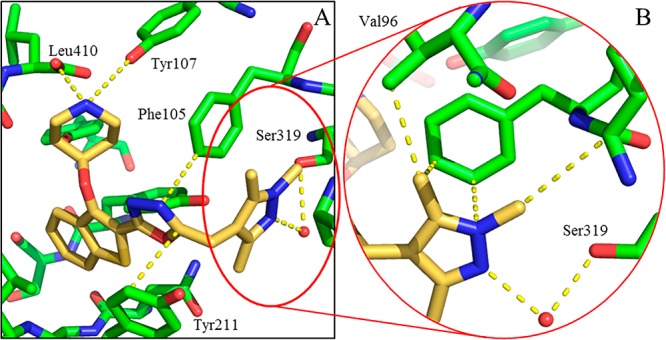
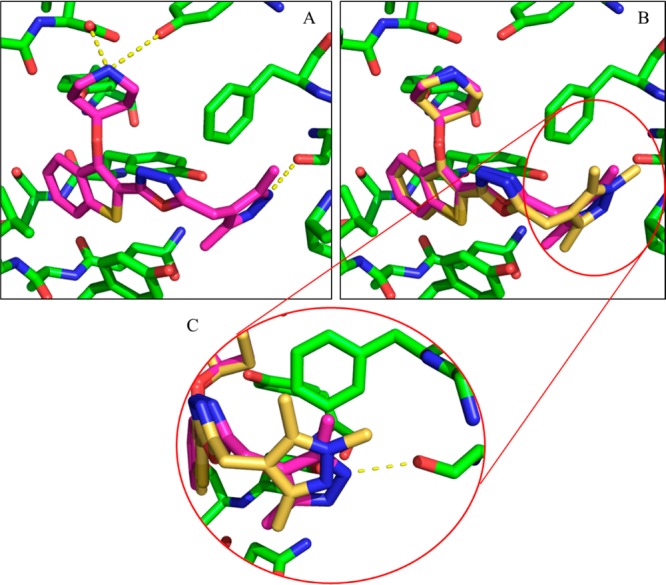
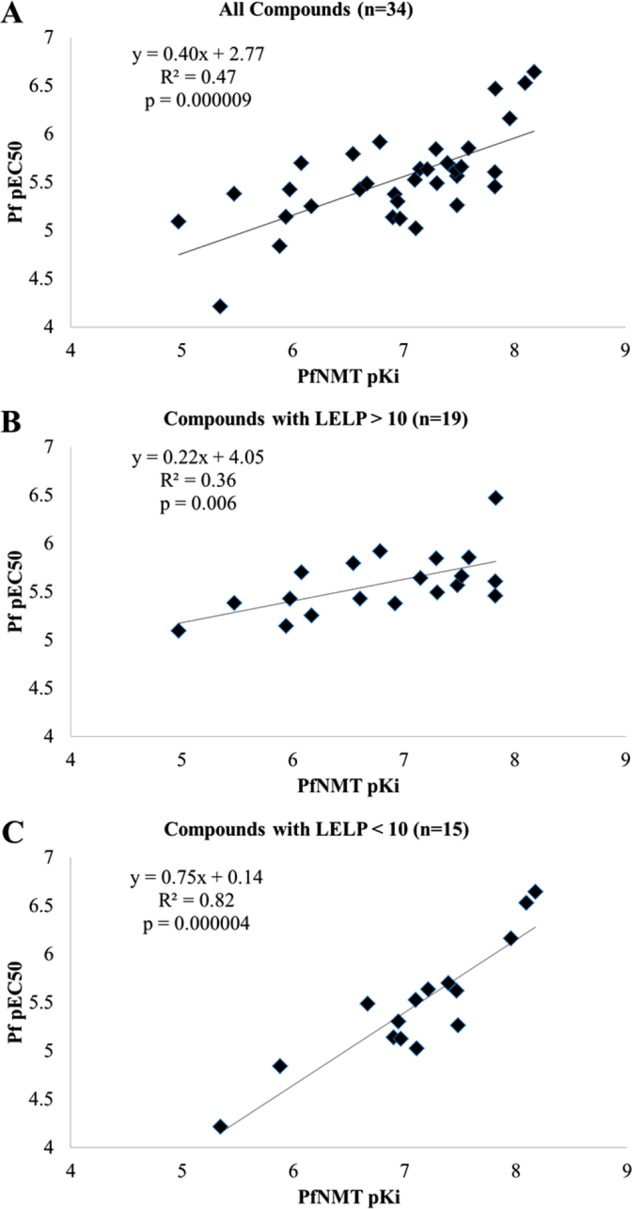
N-Myristoyltransferase (NMT) is an essential eukaryotic enzyme and an attractive drug target in parasitic infections such as malaria. We have previously reported that 2-(3-(piperidin-4-yloxy)benzo[b]thiophen-2-yl)-5-((1,3,5-trimethyl-1H-pyrazol-4-yl)methyl)-1,3,4-oxadiazole (34c) is a high affinity inhibitor of both Plasmodium falciparum and P. vivax NMT and displays activity in vivo against a rodent malaria model. Here we describe the discovery of 34c through optimization of a previously described series. Development, guided by targeting a ligand efficiency dependent lipophilicity (LELP) score of less than 10, yielded a 100-fold increase in enzyme affinity and a 100-fold drop in lipophilicity with the addition of only two heavy atoms. 34c was found to be equipotent on chloroquine-sensitive and -resistant cell lines and on both blood and liver stage forms of the parasite. These data further validate NMT as an exciting drug target in malaria and support 34c as an attractive tool for further optimization.
Figures
Similar articles
-
Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferase.J Med Chem. 2013 Jan 10;56(1):371-5. doi: 10.1021/jm301474t. Epub 2012 Dec 17. J Med Chem. 2013. PMID: 23170970 Free PMC article.
-
Design and synthesis of inhibitors of Plasmodium falciparum N-myristoyltransferase, a promising target for antimalarial drug discovery.J Med Chem. 2012 Oct 25;55(20):8879-90. doi: 10.1021/jm301160h. Epub 2012 Oct 15. J Med Chem. 2012. PMID: 23035716 Free PMC article.
-
Identification of Selective Inhibitors of Plasmodium N-Myristoyltransferase by High-Throughput Screening.J Med Chem. 2020 Jan 23;63(2):591-600. doi: 10.1021/acs.jmedchem.9b01343. Epub 2020 Jan 8. J Med Chem. 2020. PMID: 31850752
-
Current trends to design antimalarial drugs targeting N-myristoyltransferase.Future Microbiol. 2024;19(18):1601-1618. doi: 10.1080/17460913.2024.2412397. Epub 2024 Oct 23. Future Microbiol. 2024. PMID: 39440556 Review.
-
Molecular and biological aspects of antimalarial resistance in Plasmodium falciparum and Plasmodium vivax.Curr Drug Targets. 2009 Mar;10(3):279-90. doi: 10.2174/138945009787581131. Curr Drug Targets. 2009. PMID: 19275564 Review.
Cited by
-
Molecular Characterization and the Essential Biological Function of the Metal Chaperone Protein MtmA in Aspergillus fumigatus.Appl Environ Microbiol. 2022 May 10;88(9):e0018222. doi: 10.1128/aem.00182-22. Epub 2022 Apr 18. Appl Environ Microbiol. 2022. PMID: 35435716 Free PMC article.
-
Global profiling of protein lipidation using chemical proteomic technologies.Curr Opin Chem Biol. 2015 Feb;24:48-57. doi: 10.1016/j.cbpa.2014.10.016. Epub 2014 Nov 15. Curr Opin Chem Biol. 2015. PMID: 25461723 Free PMC article. Review.
-
Inhibition of protein N-myristoylation blocks Plasmodium falciparum intraerythrocytic development, egress and invasion.PLoS Biol. 2021 Oct 25;19(10):e3001408. doi: 10.1371/journal.pbio.3001408. eCollection 2021 Oct. PLoS Biol. 2021. PMID: 34695132 Free PMC article.
-
A Molecular Hybridization Approach for the Design of Potent, Highly Selective, and Brain-Penetrant N-Myristoyltransferase Inhibitors.J Med Chem. 2018 Sep 27;61(18):8374-8389. doi: 10.1021/acs.jmedchem.8b00884. Epub 2018 Sep 12. J Med Chem. 2018. PMID: 30207721 Free PMC article.
-
Ternary structure of Plasmodium vivaxN-myristoyltransferase with myristoyl-CoA and inhibitor IMP-0001173.Acta Crystallogr F Struct Biol Commun. 2024 Oct 1;80(Pt 10):269-277. doi: 10.1107/S2053230X24008604. Epub 2024 Sep 18. Acta Crystallogr F Struct Biol Commun. 2024. PMID: 39291304 Free PMC article.
References
-
- Murray C. J. L.; Rosenfeld L. C.; Lim S. S.; Andrews K. G.; Foreman K. J.; Haring D.; Fullman N.; Naghavi M.; Lozano R.; Lopez A. D. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 2012, 379, 413–431. - PubMed
-
- World Malaria Report 2011. http://www.who.int/malaria/world_malaria_report_2011/en/ (accessed Jan 18, 2012).
-
- Singh B.; Sung L. K.; Matusop A.; Radhakrishnan A.; Shamsul S. S. G.; Cox-Singh J.; Thomas A.; Conway D. J. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004, 363, 1017–1024. - PubMed
-
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M. C.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C. S.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T.-g.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334, 1372–1377. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
- G0900278/MRC_/Medical Research Council/United Kingdom
- 0900278/MRC_/Medical Research Council/United Kingdom
- MC_U117532067/MRC_/Medical Research Council/United Kingdom
- BB/D02014X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- U117532067/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Molecular Biology Databases
