

Prostate cancer ETS rearrangements switch a cell migration gene expression program from RAS/ERK to PI3K/AKT regulation
- PMID: 24642271
- PMCID: PMC3999933
- DOI: 10.1186/1476-4598-13-61
Prostate cancer ETS rearrangements switch a cell migration gene expression program from RAS/ERK to PI3K/AKT regulation
Abstract
Background: The RAS/ERK and PI3K/AKT pathways induce oncogenic gene expression programs and are commonly activated together in cancer cells. Often, RAS/ERK signaling is activated by mutation of the RAS or RAF oncogenes, and PI3K/AKT is activated by loss of the tumor suppressor PTEN. In prostate cancer, PTEN deletions are common, but, unlike other carcinomas, RAS and RAF mutations are rare. We have previously shown that over-expression of "oncogenic" ETS transcription factors, which occurs in about one-half of prostate tumors due to chromosome rearrangement, can bypass the need for RAS/ERK signaling in the activation of a cell migration gene expression program. In this study we test the role of RAS/ERK and PI3K/AKT signaling in the function of oncogenic ETS proteins.
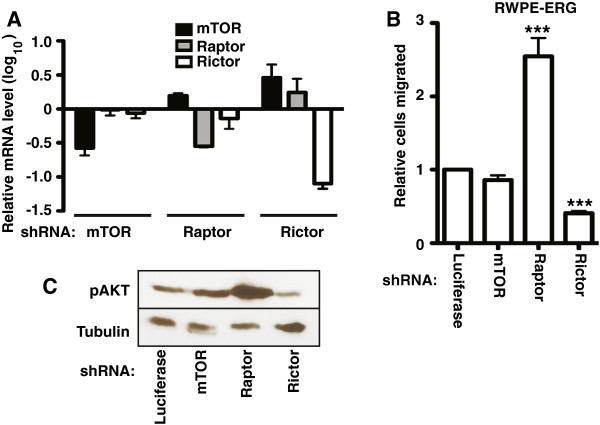
Results: We find that oncogenic ETS expression negatively correlates with RAS and RAF mutations in prostate tumors. Furthermore, the oncogenic ETS transcription factors only increased cell migration in the absence of RAS/ERK activation. In contrast to RAS/ERK, it has been reported that oncogenic ETS expression positively correlates with PI3K/AKT activation. We identified a mechanistic explanation for this finding by showing that oncogenic ETS proteins required AKT signaling to activate a cell migration gene expression program through ETS/AP-1 binding sequences. Levels of pAKT correlated with the ability of oncogenic ETS proteins to increase cell migration, but this process did not require mTORC1.
Conclusions: Our findings indicate that oncogenic ETS rearrangements cause a cell migration gene expression program to switch from RAS/ERK control to PI3K/AKT control and provide a possible explanation for the high frequency of PTEN, but not RAS/RAF mutations in prostate cancer.
Figures
Similar articles
-
RAS/ERK pathway transcriptional regulation through ETS/AP-1 binding sites.Small GTPases. 2012 Jul-Sep;3(3):154-8. doi: 10.4161/sgtp.19630. Epub 2012 Jun 1. Small GTPases. 2012. PMID: 22653334 Free PMC article.
-
PTEN expression controls cellular response to cetuximab by mediating PI3K/AKT and RAS/RAF/MAPK downstream signaling in KRAS wild-type, hormone refractory prostate cancer cells.Oncol Rep. 2009 Mar;21(3):731-5. Oncol Rep. 2009. PMID: 19212633
-
Ran is a potential therapeutic target for cancer cells with molecular changes associated with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways.Clin Cancer Res. 2012 Jan 15;18(2):380-91. doi: 10.1158/1078-0432.CCR-11-2035. Epub 2011 Nov 16. Clin Cancer Res. 2012. PMID: 22090358 Free PMC article.
-
A minireview: the role of MAPK/ERK and PI3K/Akt pathways in thyroid follicular cell-derived neoplasm.Front Biosci (Landmark Ed). 2011 Jan 1;16(2):422-39. doi: 10.2741/3696. Front Biosci (Landmark Ed). 2011. PMID: 21196179 Review.
-
Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health.Oncotarget. 2011 Mar;2(3):135-64. doi: 10.18632/oncotarget.240. Oncotarget. 2011. PMID: 21411864 Free PMC article. Review.
Cited by
-
Extracellular signal-regulated kinase signaling regulates the opposing roles of JUN family transcription factors at ETS/AP-1 sites and in cell migration.Mol Cell Biol. 2015 Jan;35(1):88-100. doi: 10.1128/MCB.00982-14. Epub 2014 Oct 20. Mol Cell Biol. 2015. PMID: 25332240 Free PMC article.
-
Suppression of CHK1 by ETS Family Members Promotes DNA Damage Response Bypass and Tumorigenesis.Cancer Discov. 2015 May;5(5):550-63. doi: 10.1158/2159-8290.CD-13-1050. Epub 2015 Feb 4. Cancer Discov. 2015. PMID: 25653093 Free PMC article.
-
Statins affect ETS1-overexpressing triple-negative breast cancer cells by restoring DUSP4 deficiency.Sci Rep. 2016 Sep 8;6:33035. doi: 10.1038/srep33035. Sci Rep. 2016. PMID: 27604655 Free PMC article.
-
Calcium signalling pathways in prostate cancer initiation and progression.Nat Rev Urol. 2023 Sep;20(9):524-543. doi: 10.1038/s41585-023-00738-x. Epub 2023 Mar 24. Nat Rev Urol. 2023. PMID: 36964408 Review.
-
Interaction with ZMYND11 mediates opposing roles of Ras-responsive transcription factors ETS1 and ETS2.Nucleic Acids Res. 2017 May 5;45(8):4452-4462. doi: 10.1093/nar/gkx039. Nucleic Acids Res. 2017. PMID: 28119415 Free PMC article.
References
-
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
