

Evolutionary conservation of the eumetazoan gene regulatory landscape
- PMID: 24642862
- PMCID: PMC3975063
- DOI: 10.1101/gr.162529.113
Evolutionary conservation of the eumetazoan gene regulatory landscape
Abstract
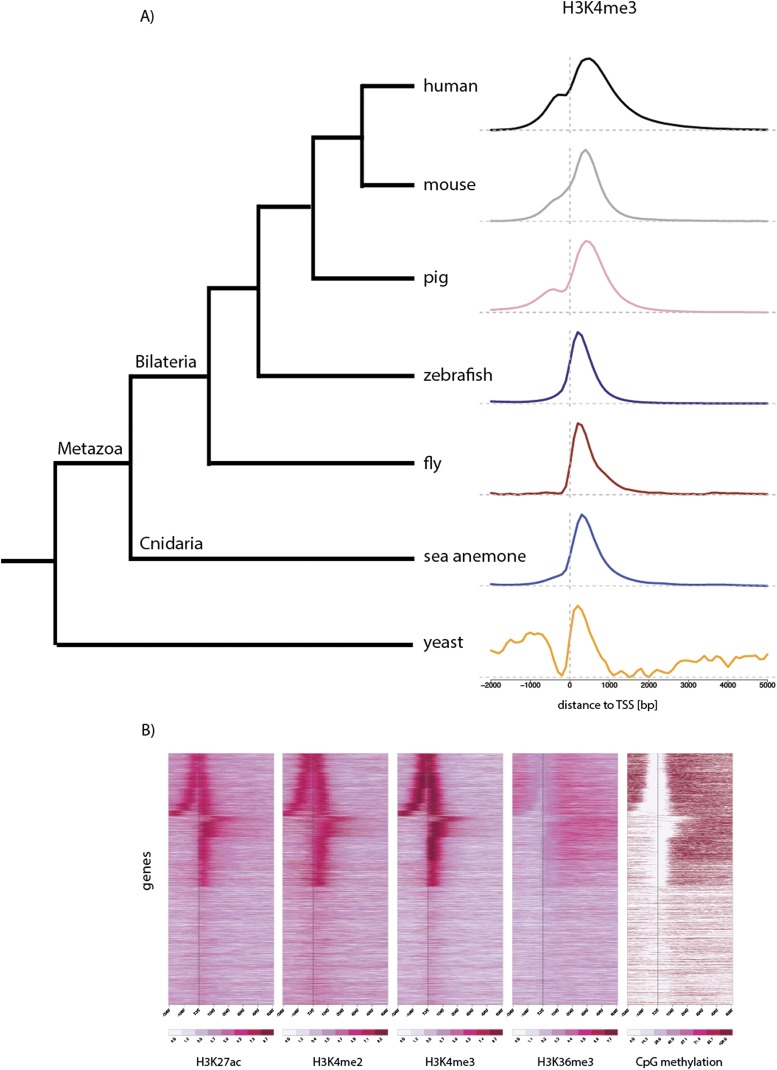
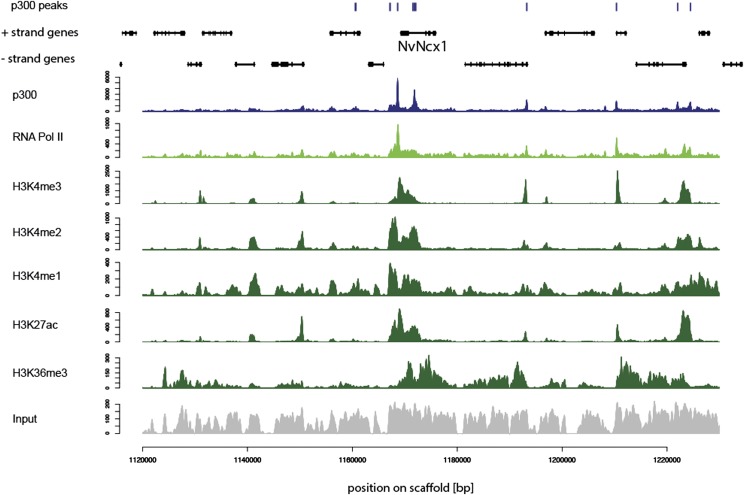
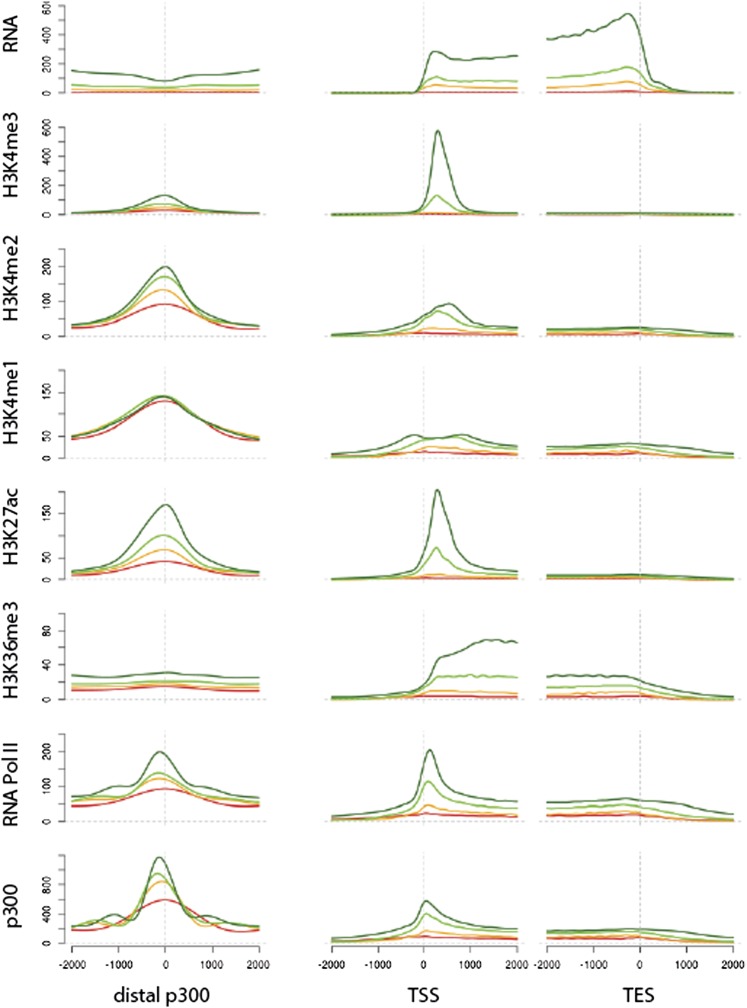
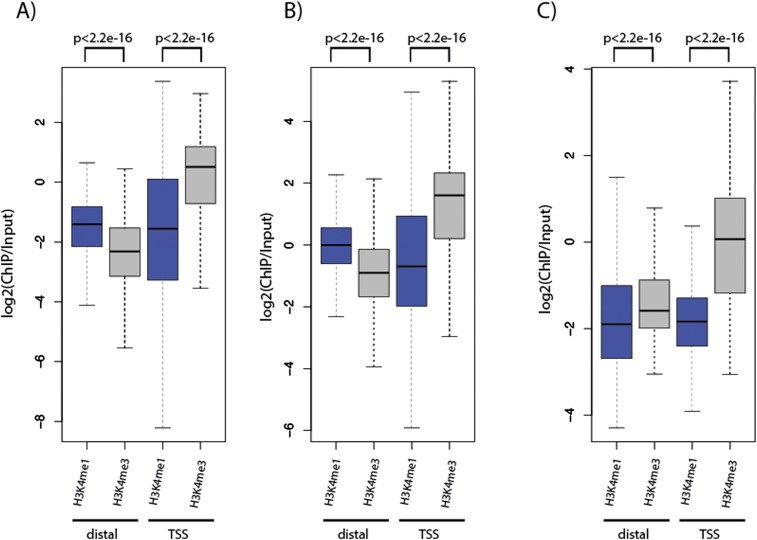
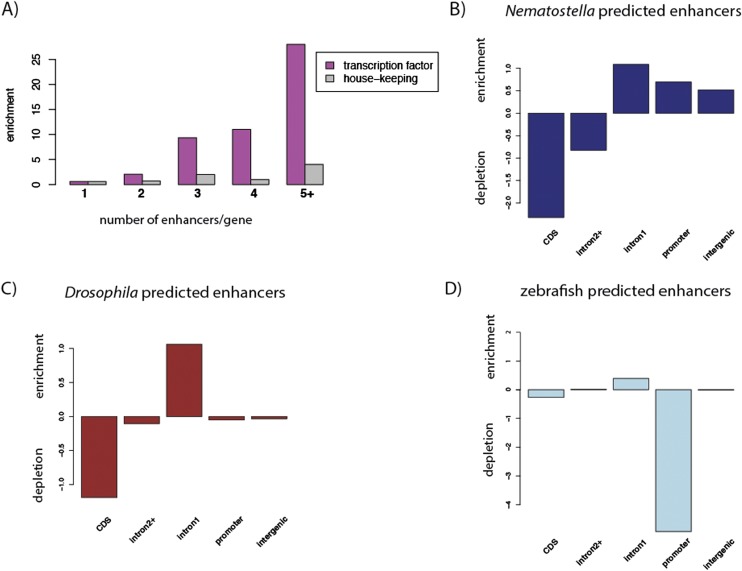
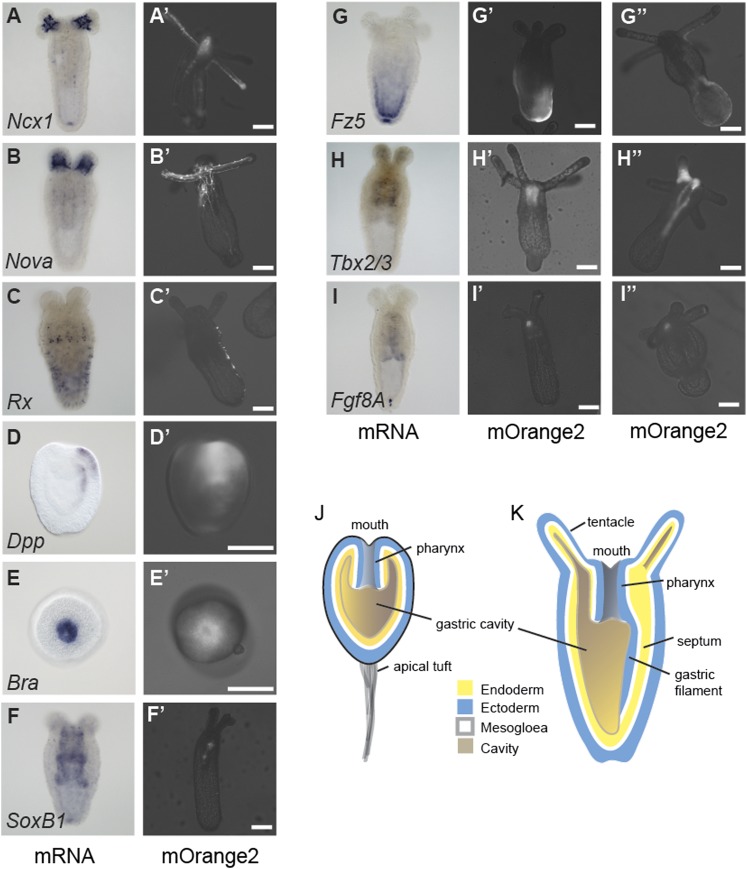
Despite considerable differences in morphology and complexity of body plans among animals, a great part of the gene set is shared among Bilateria and their basally branching sister group, the Cnidaria. This suggests that the common ancestor of eumetazoans already had a highly complex gene repertoire. At present it is therefore unclear how morphological diversification is encoded in the genome. Here we address the possibility that differences in gene regulation could contribute to the large morphological divergence between cnidarians and bilaterians. To this end, we generated the first genome-wide map of gene regulatory elements in a nonbilaterian animal, the sea anemone Nematostella vectensis. Using chromatin immunoprecipitation followed by deep sequencing of five chromatin modifications and a transcriptional cofactor, we identified over 5000 enhancers in the Nematostella genome and could validate 75% of the tested enhancers in vivo. We found that in Nematostella, but not in yeast, enhancers are characterized by the same combination of histone modifications as in bilaterians, and these enhancers preferentially target developmental regulatory genes. Surprisingly, the distribution and abundance of gene regulatory elements relative to these genes are shared between Nematostella and bilaterian model organisms. Our results suggest that complex gene regulation originated at least 600 million yr ago, predating the common ancestor of eumetazoans.
Figures
References
-
- Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339: 1074–1077 - PubMed
-
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 - PubMed
-
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D 2004. Ultraconserved elements in the human genome. Science 304: 1321–1325 - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases