

Molecular basis for increased risk for late-onset Alzheimer disease due to the naturally occurring L28P mutation in apolipoprotein E4
- PMID: 24644280
- PMCID: PMC4007480
- DOI: 10.1074/jbc.M113.538124
Molecular basis for increased risk for late-onset Alzheimer disease due to the naturally occurring L28P mutation in apolipoprotein E4
Abstract
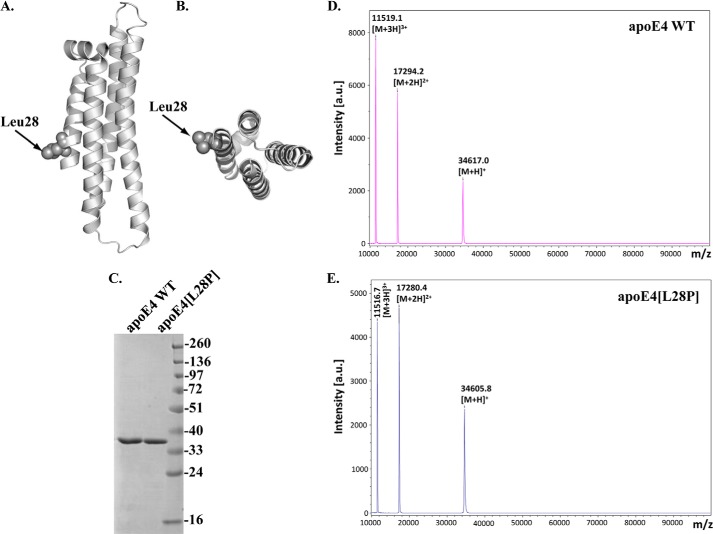
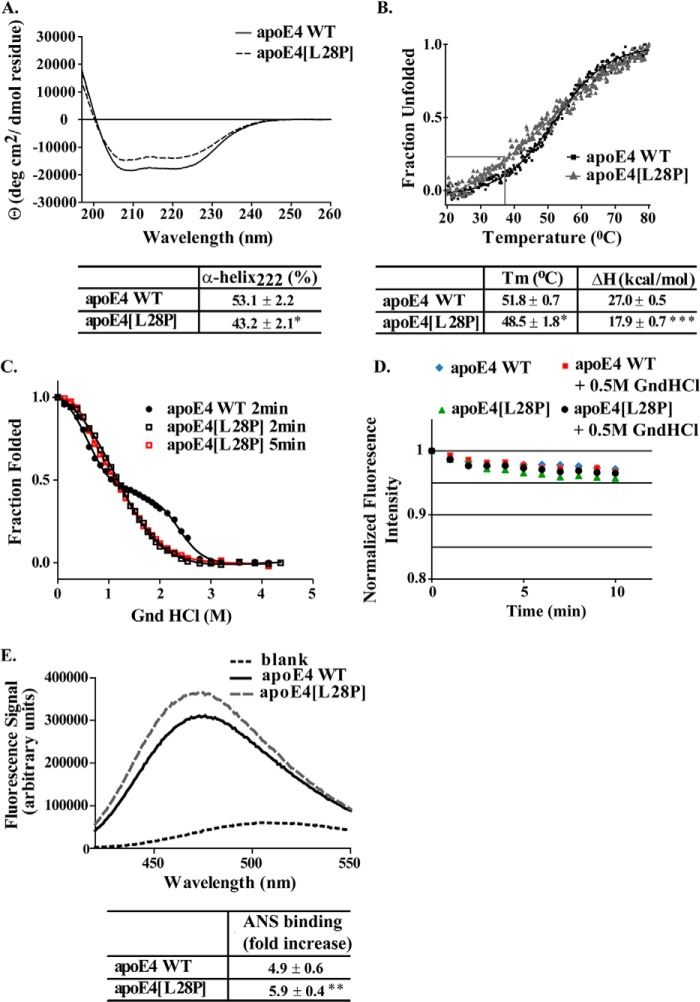
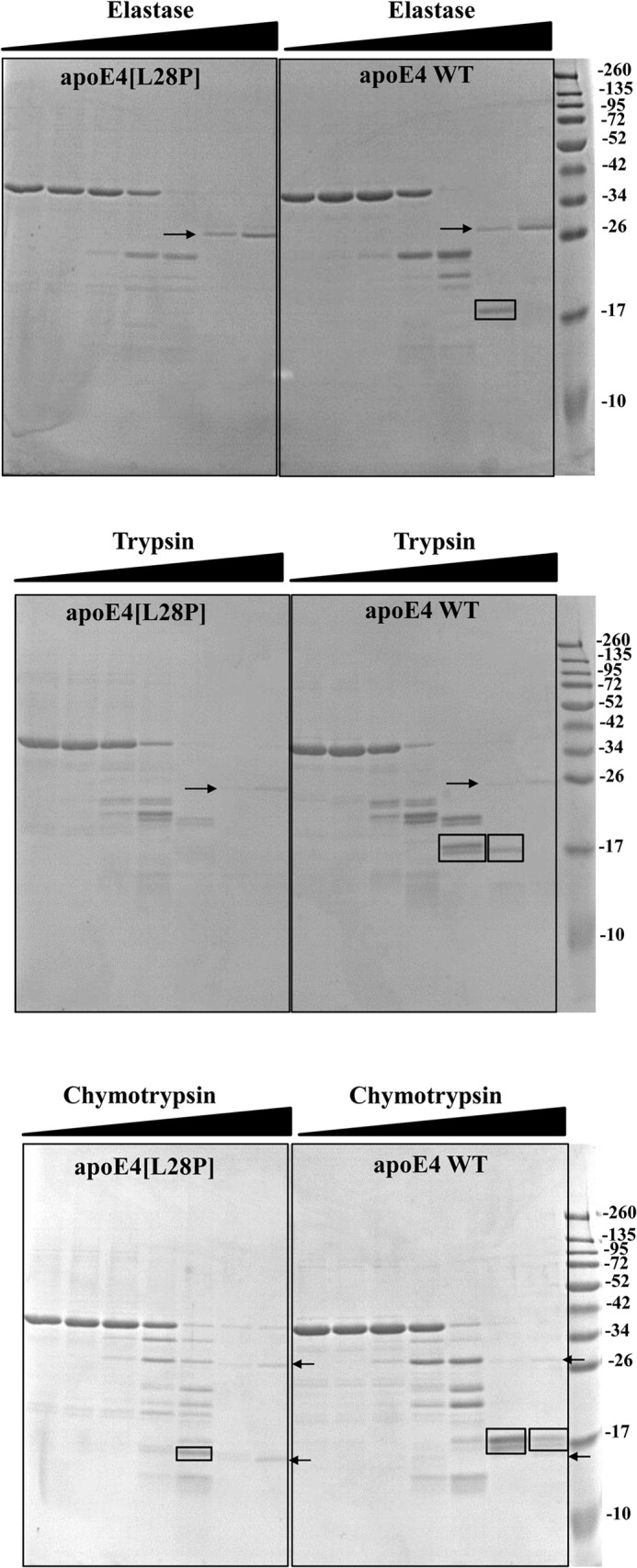
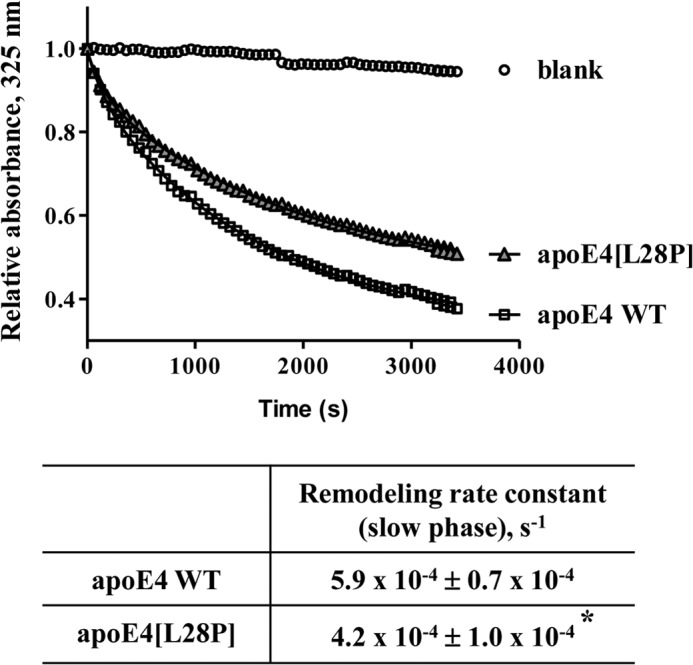
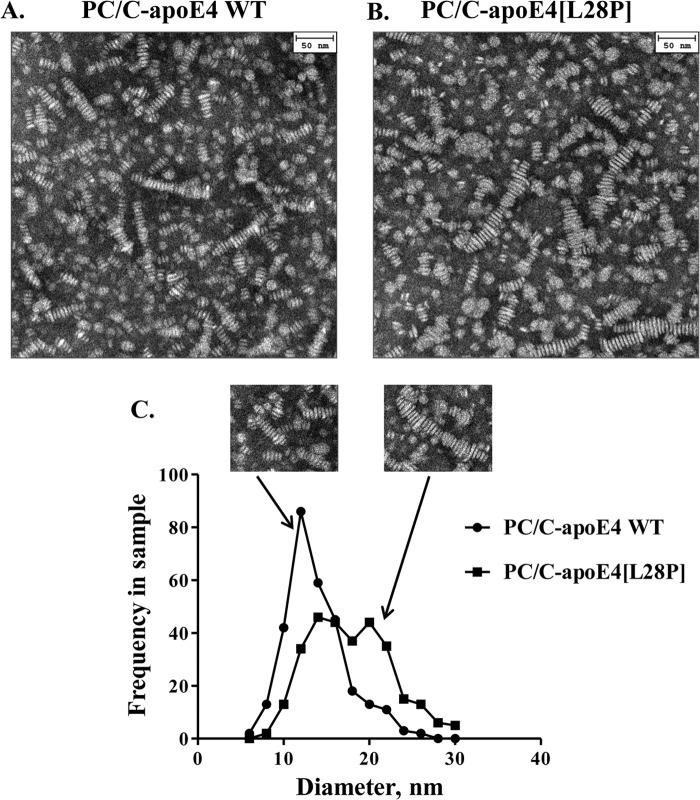
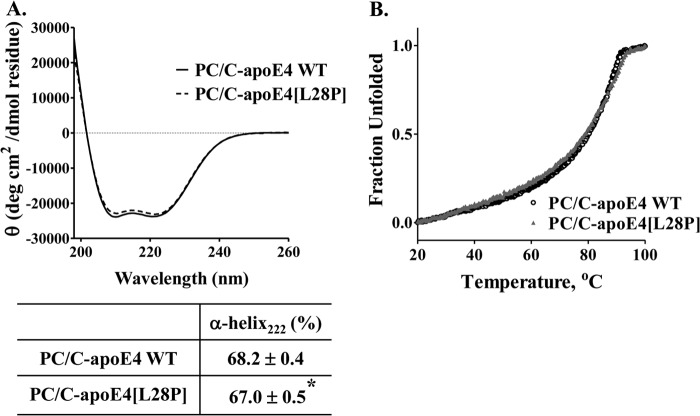
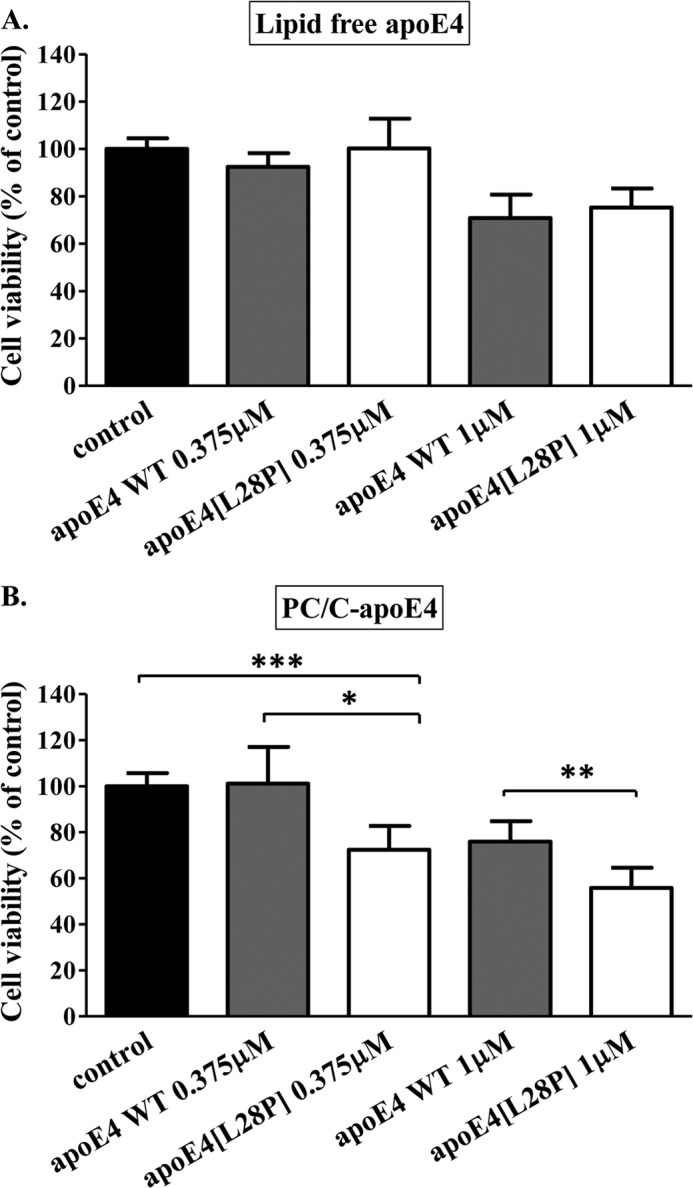
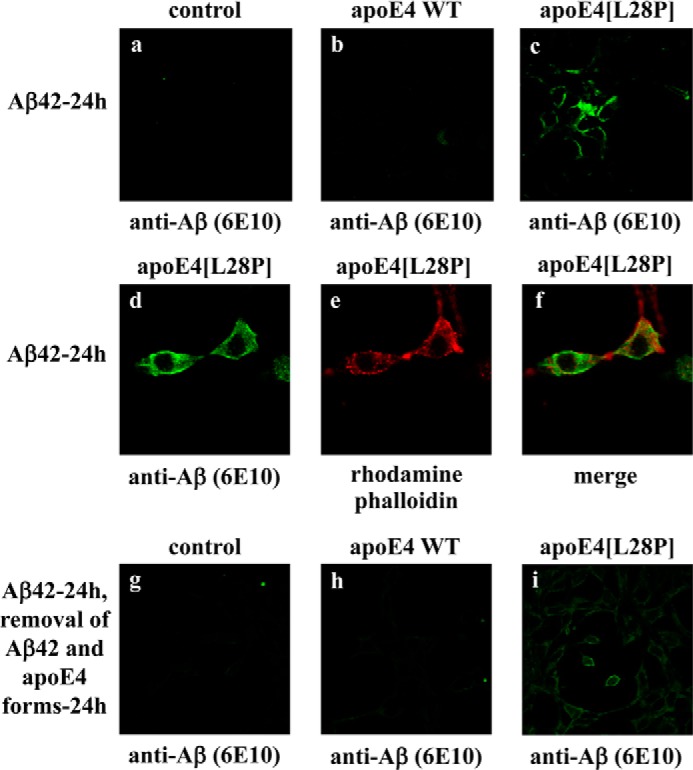
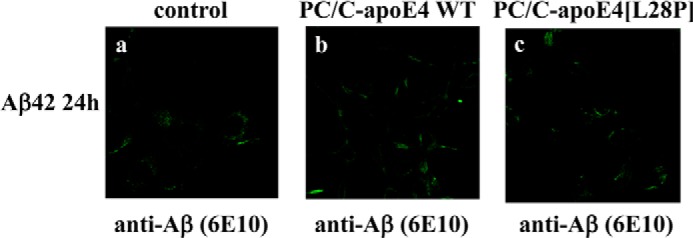
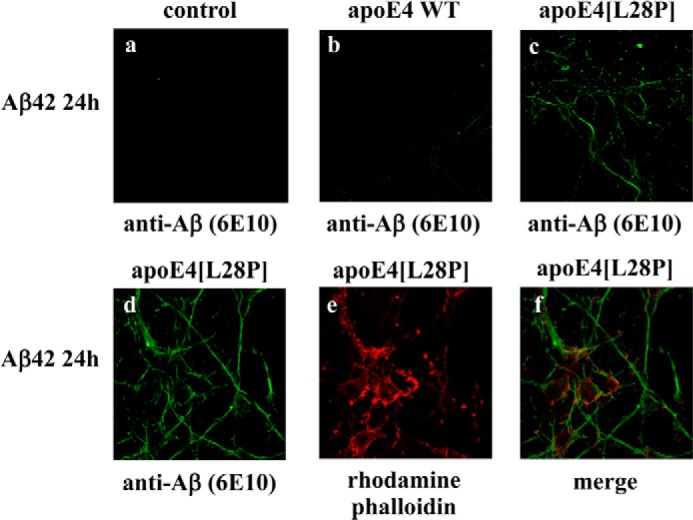
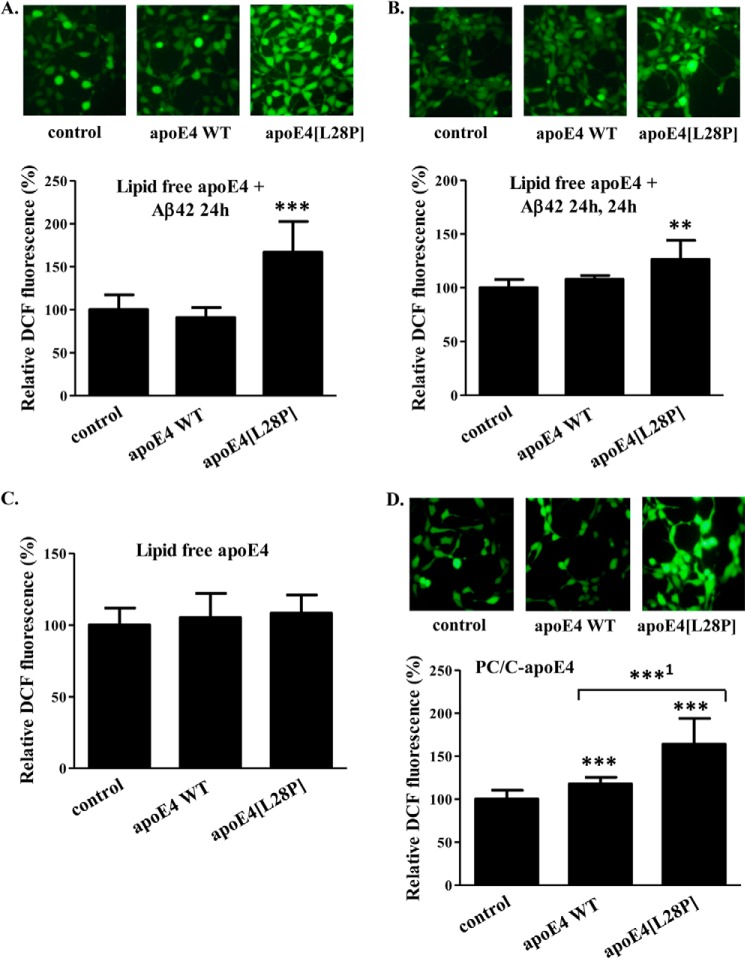
The apolipoprotein (apo) E4 isoform has consistently emerged as a susceptibility factor for late-onset Alzheimer disease (AD), although the exact mechanism is not clear. A rare apoE4 mutant, apoE4[L28P] Pittsburgh, burdens carriers with an added risk for late-onset AD and may be a useful tool for gaining insights into the role of apoE4 in disease pathogenesis. Toward this end, we evaluated the effect of the L28P mutation on the structural and functional properties of apoE4. ApoE4[L28P] was found to have significantly perturbed thermodynamic properties, to have reduced helical content, and to expose a larger portion of the hydrophobic surface to the solvent. Furthermore, this mutant is thermodynamically destabilized and more prone to proteolysis. When interacting with lipids, apoE4[L28P] formed populations of lipoprotein particles with structural defects. The structural perturbations brought about by the mutation were accompanied by aberrant functions associated with the pathogenesis of AD. Specifically, apoE4[L28P] promoted the cellular uptake of extracellular amyloid β peptide 42 (Aβ42) by human neuroblastoma SK-N-SH cells as well as by primary mouse neuronal cells and led to increased formation of intracellular reactive oxygen species that persisted for at least 24 h. Furthermore, lipoprotein particles containing apoE4[L28P] induced intracellular reactive oxygen species formation and reduced SK-N-SH cell viability. Overall, our findings suggest that the L28P mutation leads to significant structural and conformational perturbations in apoE4 and can induce functional defects associated with neuronal Aβ42 accumulation and oxidative stress. We propose that these structural and functional changes underlie the observed added risk for AD development in carriers of apoE4[L28P].
Keywords: Alzheimer Disease; Amyloid Peptide β; ApoE; Apolipoproteins; Biophysics; Lipoprotein; Mutation; Reactive Oxygen Species (ROS); Thermodynamic Stability; Toxicity.
Figures
References
-
- Zannis V. I., Kypreos K. E., Chroni A., Kardassis D., Zanni E. E. (2004) in Molecular Mechanisms of Atherosclerosis (Loscalzo J., ed) pp. 111–174, Taylor & Francis, Abington, UK
-
- Zannis V. I., Cole F. S., Jackson C. L., Kurnit D. M., Karathanasis S. K. (1985) Distribution of apolipoprotein A-I, C-II, C-III, and E mRNA in fetal human tissues: time-dependent induction of apolipoprotein E mRNA by cultures of human monocyte-macrophages. Biochemistry 24, 4450–4455 - PubMed
-
- Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 - PubMed
-
- Myers R. H., Schaefer E. J., Wilson P. W., D'Agostino R., Ordovas J. M., Espino A., Au R., White R. F., Knoefel J. E., Cobb J. L., McNulty K. A., Beiser A., Wolf P. A. (1996) Apolipoprotein E ϵ4 association with dementia in a population-based study: The Framingham study. Neurology 46, 673–677 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
