

Whole-genome sequencing of tibetan macaque (Macaca Thibetana) provides new insight into the macaque evolutionary history
- PMID: 24648498
- PMCID: PMC4032132
- DOI: 10.1093/molbev/msu104
Whole-genome sequencing of tibetan macaque (Macaca Thibetana) provides new insight into the macaque evolutionary history
Abstract
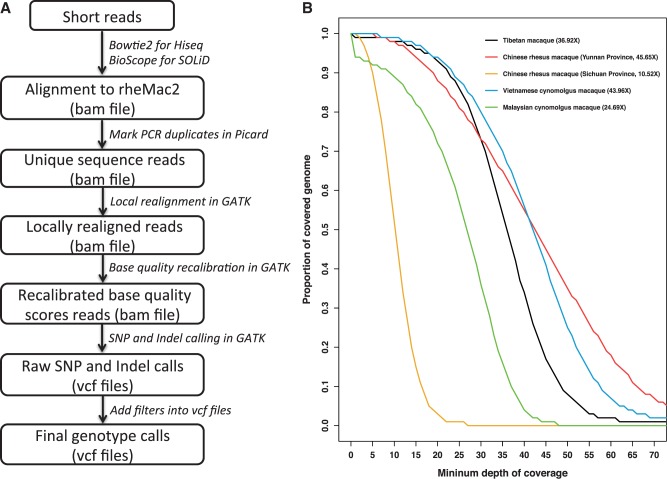
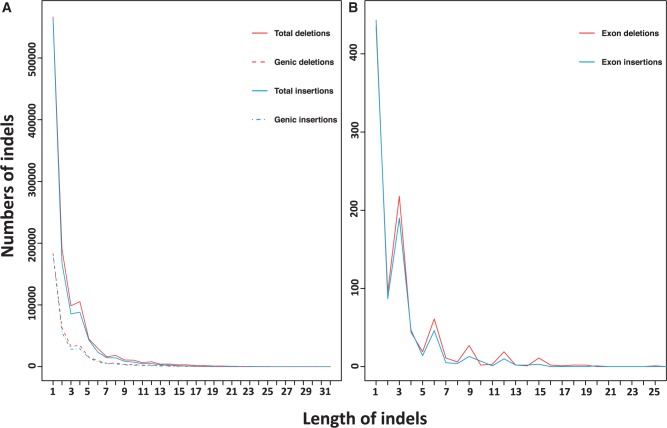
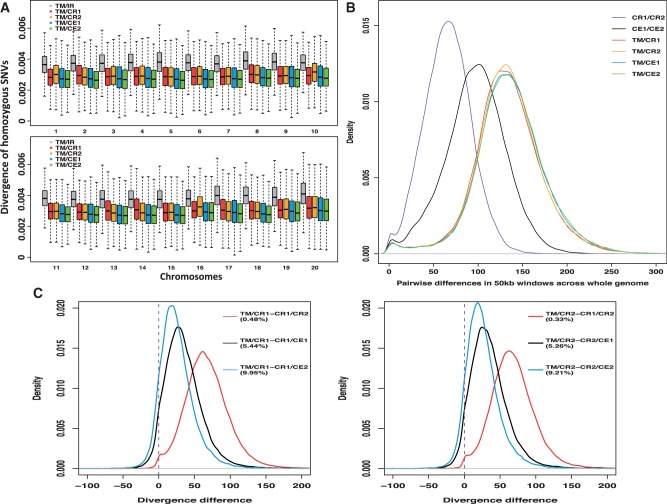
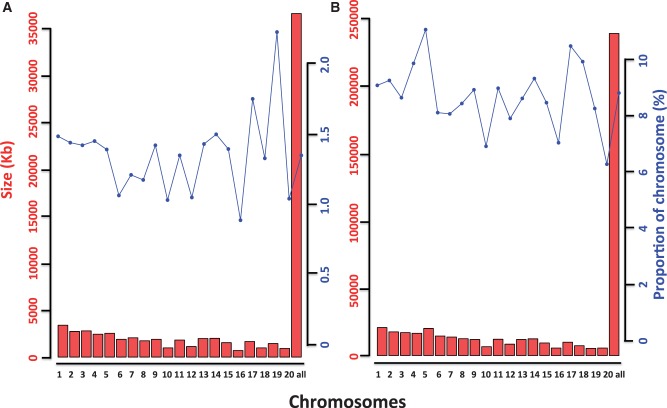
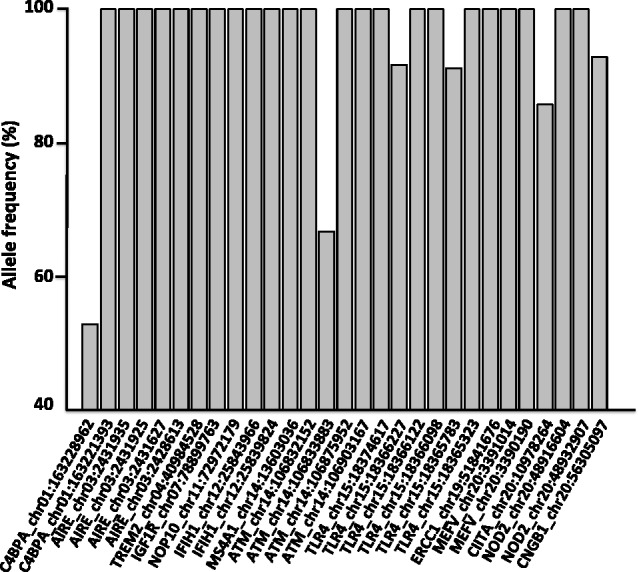
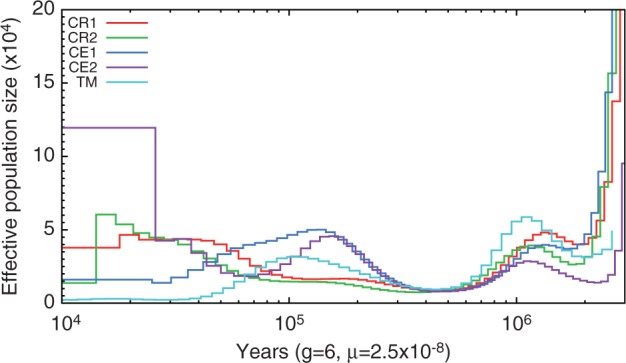
Macaques are the most widely distributed nonhuman primates and used as animal models in biomedical research. The availability of full-genome sequences from them would be essential to both biomedical and primate evolutionary studies. Previous studies have reported whole-genome sequences from rhesus macaque (Macaca mulatta) and cynomolgus macaque (M. fascicularis, CE), both of which belong to the fascicularis group. Here, we present a 37-fold coverage genome sequence of the Tibetan macaque (M. thibetana; TM). TM is an endemic species to China belonging to the sinica group. On the basis of mapping to the rhesus macaque genome, we identified approximately 11.9 million single-nucleotide variants), of which 3.9 million were TM specific, as assessed by comparison two Chinese rhesus macaques (CR) and two CE genomes. Some genes carried TM-specific homozygous nonsynonymous variants (TSHNVs), which were scored as deleterious in human by both PolyPhen-2 and SIFT (Sorting Tolerant From Intolerant) and were enriched in the eye disease genes. In total, 273 immune response and disease-related genes carried at least one TSHNV. The heterozygosity rates of two CRs (0.002617 and 0.002612) and two CEs (0.003004 and 0.003179) were approximately three times higher than that of TM (0.000898). Polymerase chain reaction resequencing of 18 TM individuals showed that 29 TSHNVs exhibited high allele frequencies, thus confirming their low heterozygosity. Genome-wide genetic divergence analysis demonstrated that TM was more closely related to CR than to CE. We further detected unusual low divergence regions between TM and CR. In addition, after applying statistical criteria to detect putative introgression regions (PIRs) in the TM genome, up to 239,620 kb PIRs (8.84% of the genome) were identified. Given that TM and CR have overlapping geographical distributions, had the same refuge during the Middle Pleistocene, and show similar mating behaviors, it is highly likely that there was an ancient introgression event between them. Moreover, demographic inferences revealed that TM exhibited a similar demographic history as other macaques until 0.5 Ma, but then it maintained a lower effective population size until present time. Our study has provided new insight into the macaque evolutionary history, confirming hybridization events between macaque species groups based on genome-wide data.
Keywords: SNVs; Tibetan macaque; demographic trajectories; genetic divergence; introgression; whole-genome sequencing.
© The Author 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
Similar articles
-
Whole-genome sequencing and analysis of the Malaysian cynomolgus macaque (Macaca fascicularis) genome.Genome Biol. 2012 Jul 2;13(7):R58. doi: 10.1186/gb-2012-13-7-r58. Genome Biol. 2012. PMID: 22747675 Free PMC article.
-
Ancient hybridization and admixture in macaques (genus Macaca) inferred from whole genome sequences.Mol Phylogenet Evol. 2018 Oct;127:376-386. doi: 10.1016/j.ympev.2018.03.038. Epub 2018 Mar 31. Mol Phylogenet Evol. 2018. PMID: 29614345
-
Genomic Evidence for the Complex Evolutionary History of Macaques (Genus Macaca).J Mol Evol. 2024 Jun;92(3):286-299. doi: 10.1007/s00239-024-10166-z. Epub 2024 Apr 18. J Mol Evol. 2024. PMID: 38634872
-
Haplessly hoping: macaque major histocompatibility complex made easy.ILAR J. 2013;54(2):196-210. doi: 10.1093/ilar/ilt036. ILAR J. 2013. PMID: 24174442 Free PMC article. Review.
-
Nonhuman primate models in the genomic era: a paradigm shift.ILAR J. 2013;54(2):154-65. doi: 10.1093/ilar/ilt044. ILAR J. 2013. PMID: 24174439 Free PMC article. Review.
Cited by
-
Evolution of genes involved in the unusual genitals of the bear macaque, Macaca arctoides.Ecol Evol. 2022 May 24;12(5):e8897. doi: 10.1002/ece3.8897. eCollection 2022 May. Ecol Evol. 2022. PMID: 35646310 Free PMC article.
-
Comparative genomic analysis of sifakas (Propithecus) reveals selection for folivory and high heterozygosity despite endangered status.Sci Adv. 2021 Apr 23;7(17):eabd2274. doi: 10.1126/sciadv.abd2274. Print 2021 Apr. Sci Adv. 2021. PMID: 33893095 Free PMC article.
-
Emergence and evolution of inter-specific segregating retrocopies in cynomolgus monkey (Macaca fascicularis) and rhesus macaque (Macaca mulatta).Sci Rep. 2016 Sep 7;6:32598. doi: 10.1038/srep32598. Sci Rep. 2016. PMID: 27600022 Free PMC article.
-
The rhesus macaque as a success story of the Anthropocene.Elife. 2022 Jul 8;11:e78169. doi: 10.7554/eLife.78169. Elife. 2022. PMID: 35801697 Free PMC article.
-
Comparison of sequencing data processing pipelines and application to underrepresented African human populations.BMC Bioinformatics. 2021 Oct 9;22(1):488. doi: 10.1186/s12859-021-04407-x. BMC Bioinformatics. 2021. PMID: 34627144 Free PMC article.
References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300.
-
- Berard J. A four-year study of the association between male dominance rank, residency status, and reproductive activity in rhesus macaques (Macaca mulatta) Primates. 1999;40:159–175. - PubMed
-
- Blancher A, Bonhomme M, Crouau-Roy B, Terao K, Kitano T, Saitou N. Mitochondrial DNA sequence phylogeny of 4 populations of the widely distributed cynomolgus macaque (Macaca fascicularis fascicularis) J Hered. 2008;99:254–264. - PubMed
-
- Bonhomme M, Cuartero S, Blancher A, Crouau-Roy B. Assessing natural introgression in 2 biomedical model species, the rhesus macaque (Macaca mulatta) and the long-tailed macaque (Macaca fascicularis) J Hered. 2009;100:158–169. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
