

The characteristic direction: a geometrical approach to identify differentially expressed genes
- PMID: 24650281
- PMCID: PMC4000056
- DOI: 10.1186/1471-2105-15-79
The characteristic direction: a geometrical approach to identify differentially expressed genes
Abstract
Background: Identifying differentially expressed genes (DEG) is a fundamental step in studies that perform genome wide expression profiling. Typically, DEG are identified by univariate approaches such as Significance Analysis of Microarrays (SAM) or Linear Models for Microarray Data (LIMMA) for processing cDNA microarrays, and differential gene expression analysis based on the negative binomial distribution (DESeq) or Empirical analysis of Digital Gene Expression data in R (edgeR) for RNA-seq profiling.
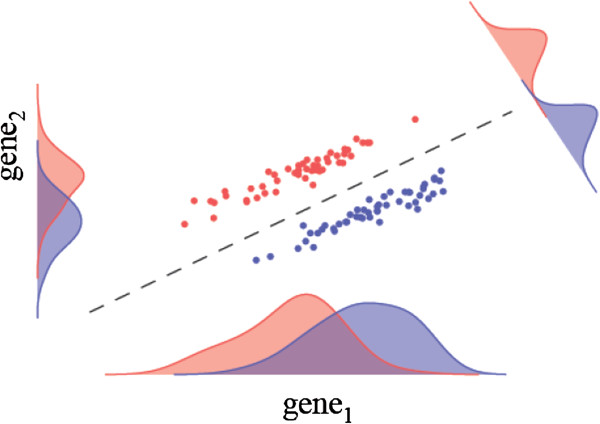
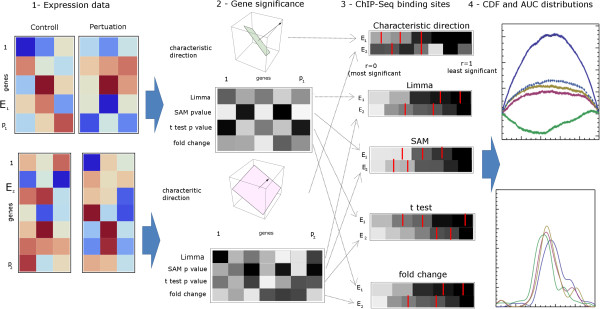
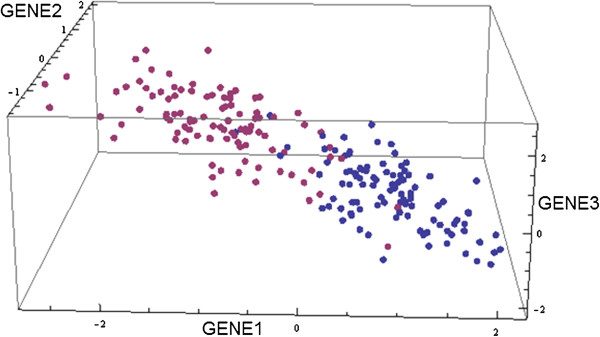
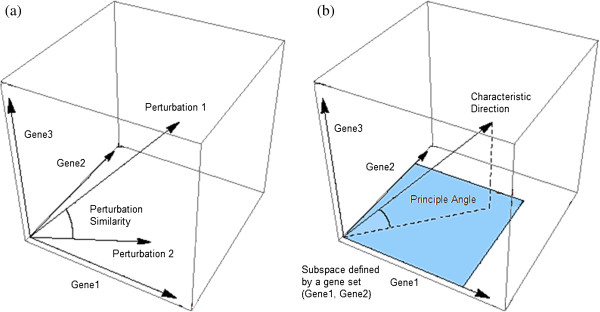
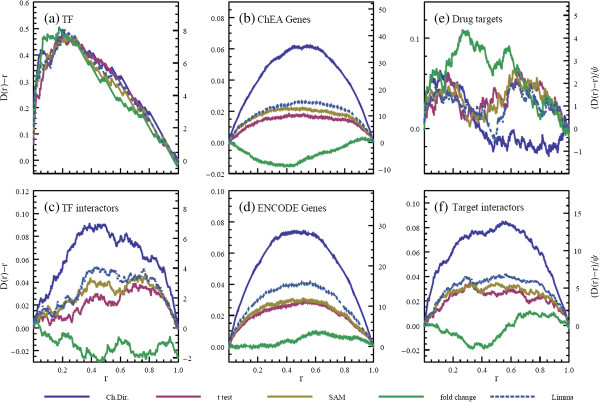
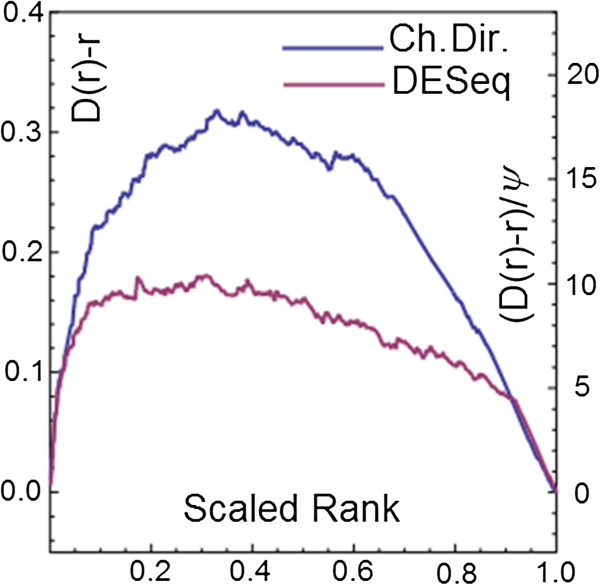
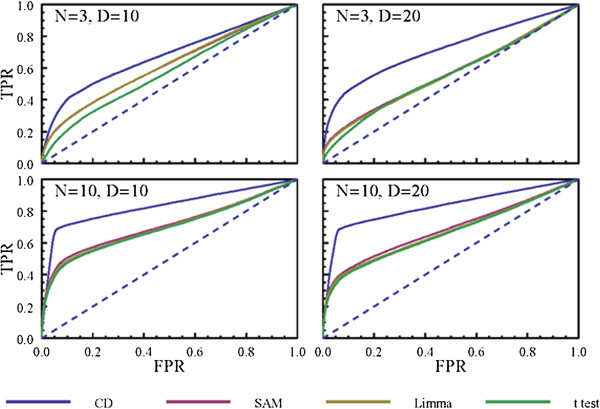
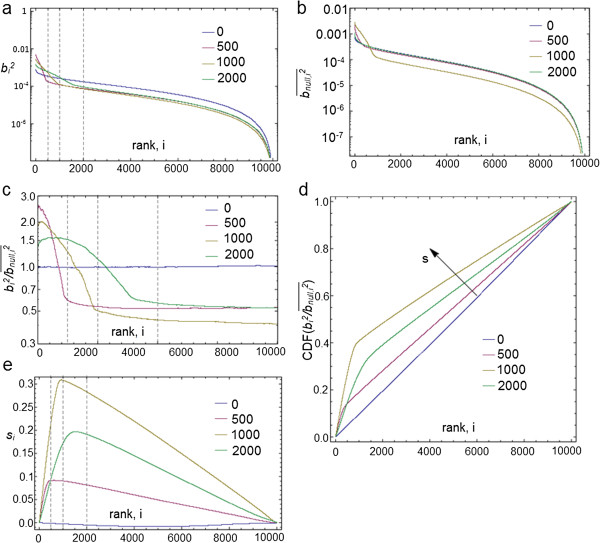
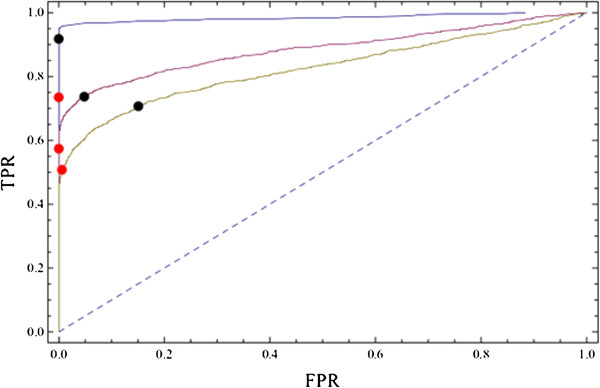
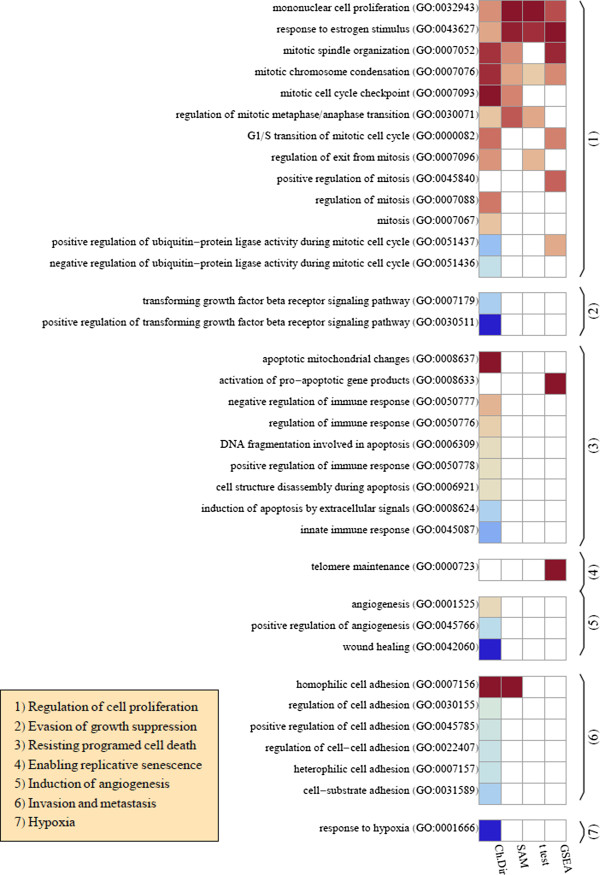
Results: Here we present a new geometrical multivariate approach to identify DEG called the Characteristic Direction. We demonstrate that the Characteristic Direction method is significantly more sensitive than existing methods for identifying DEG in the context of transcription factor (TF) and drug perturbation responses over a large number of microarray experiments. We also benchmarked the Characteristic Direction method using synthetic data, as well as RNA-Seq data. A large collection of microarray expression data from TF perturbations (73 experiments) and drug perturbations (130 experiments) extracted from the Gene Expression Omnibus (GEO), as well as an RNA-Seq study that profiled genome-wide gene expression and STAT3 DNA binding in two subtypes of diffuse large B-cell Lymphoma, were used for benchmarking the method using real data. ChIP-Seq data identifying DNA binding sites of the perturbed TFs, as well as known drug targets of the perturbing drugs, were used as prior knowledge silver-standard for validation. In all cases the Characteristic Direction DEG calling method outperformed other methods. We find that when drugs are applied to cells in various contexts, the proteins that interact with the drug-targets are differentially expressed and more of the corresponding genes are discovered by the Characteristic Direction method. In addition, we show that the Characteristic Direction conceptualization can be used to perform improved gene set enrichment analyses when compared with the gene-set enrichment analysis (GSEA) and the hypergeometric test.
Conclusions: The application of the Characteristic Direction method may shed new light on relevant biological mechanisms that would have remained undiscovered by the current state-of-the-art DEG methods. The method is freely accessible via various open source code implementations using four popular programming languages: R, Python, MATLAB and Mathematica, all available at: http://www.maayanlab.net/CD.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
