

Comment
doi: 10.1038/nchem.1898.
Contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein
Affiliations
- PMID: 24651178
- PMCID: PMC4217633
- DOI: 10.1038/nchem.1898
Item in Clipboard
Comment
Contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein
Nat Chem.
2014 Apr.
No abstract available
Figures
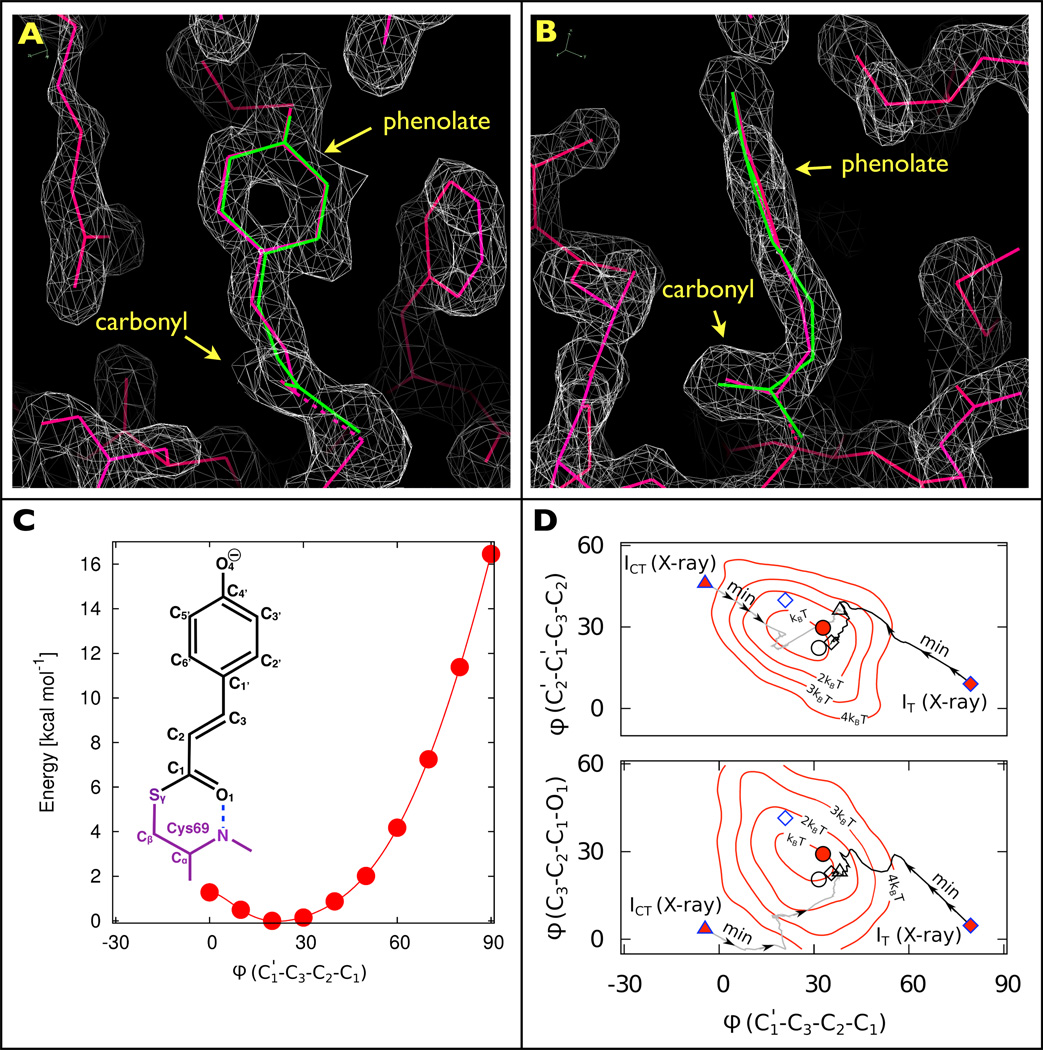
Structures and stereochemistry of early intermediates following photoactivation of PYP. a, Front and b, side views of the pCA chromophore and its immediate surroundings. The two electron density maps for IT (grey), contoured at 1.64σ, clearly show the carbonyl oriented perpendicular to the phenolate plane. Although quite different stereochemically, both X-ray (red) and DFT (green) pCA structures thread through the electron density maps with high fidelity. c, Labelling scheme for the pCA chromophore and DFT energies (calculated at the D-B3LYP/def2-TZVP level) for the C3=C2 dihedral angle (ϕ) of pR0. d, Dihedral angles of early intermediates reported by Jung et al. (blue outline: IT as diamonds; ICT as triangles), and Schotte et al. (black outline: pR0 as circles), for both X-ray refined structures (red-filled symbols) and DFT-optimized structures (open symbols). The X-ray structures for IT and ICT were found to be unstable: during DFT structure optimization, both converged to structures similar to pR0 (the black and grey lines, labelled ‘Min’, indicate the dihedral projections along the energy minimization pathways obtained during the respective structure optimizations). The DFT calculations reported in Schotte et al. included 176 atoms (D-BP86/def2-SVP); Jung et al. included 157 atoms (B97-1/6-31G(d)/3-21G). Underlaid are dihedral/dihedral free-energy contours computed from a 5 ps hybrid quantum/classical mechanics (QM/MM) simulation, of the pR0 intermediate with 143 QM atoms/2,171 MM atoms (simulations were performed using CHARMM/Q-Chem: D-BP86/def2-SVP/CHARMM27). Note that in this projection, the Jung et al. DFT structure for IT differs from the IT, ICT and pR0 DFT-optimized cluster by only ~1 kT in free energy.
Comment in
-
Reply to 'contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein'.Nat Chem. 2014 Apr;6(4):259-60. doi: 10.1038/nchem.1897. Nat Chem. 2014. PMID: 24651179 Free PMC article. No abstract available.
Comment on
-
Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography.Nat Chem. 2013 Mar;5(3):212-20. doi: 10.1038/nchem.1565. Epub 2013 Feb 3. Nat Chem. 2013. PMID: 23422563 Free PMC article.
References
-
- Groenhof G, et al. J. Am. Chem. Soc. 2004;126:4228–4233. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
