

Physics of cell adhesion: some lessons from cell-mimetic systems
- PMID: 24651316
- PMCID: PMC4028615
- DOI: 10.1039/c3sm51910d
Physics of cell adhesion: some lessons from cell-mimetic systems
Abstract
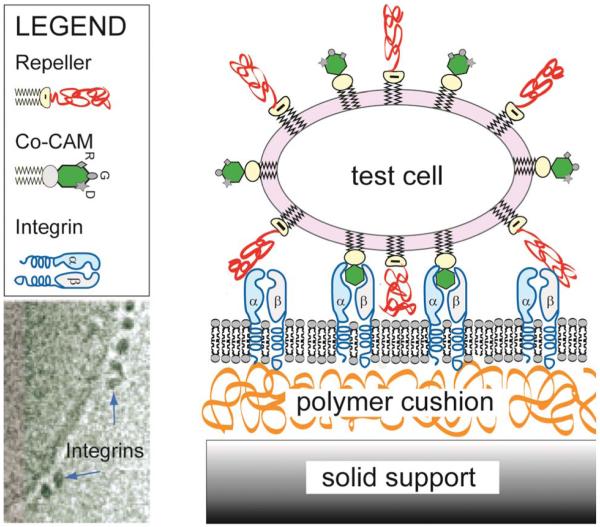
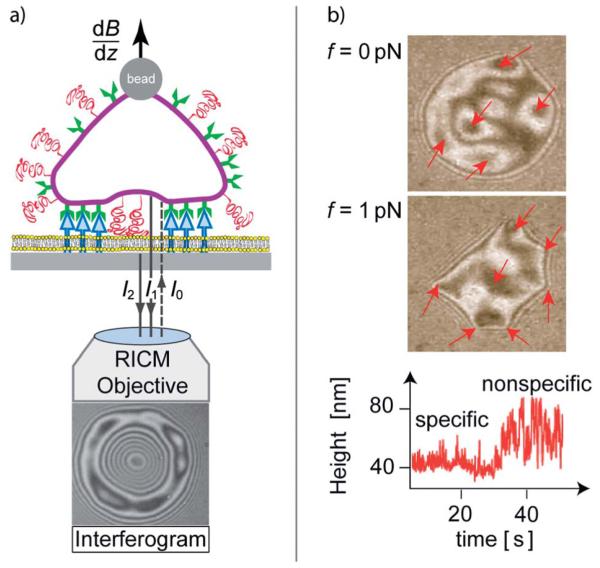
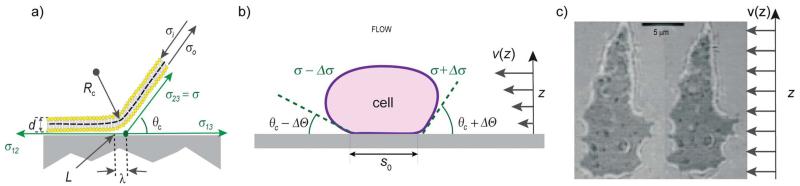
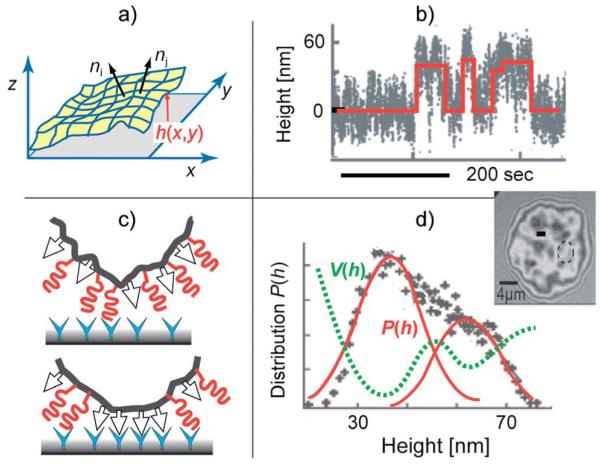
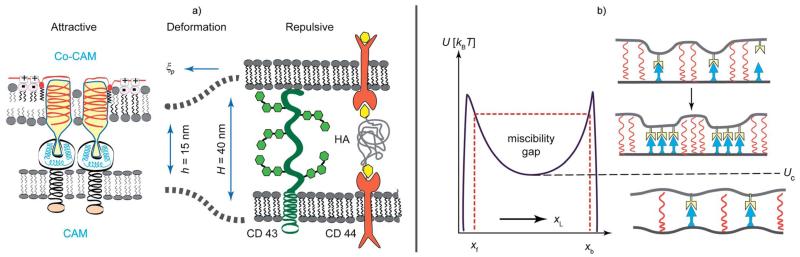
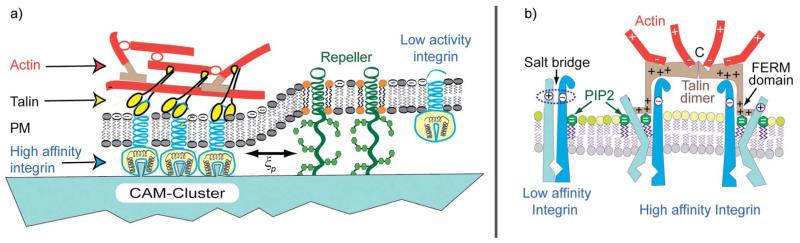
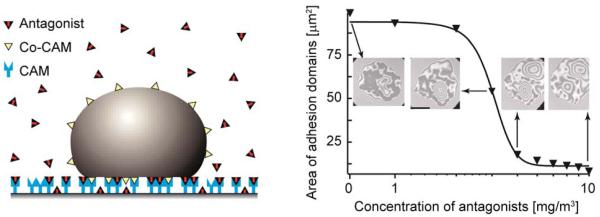
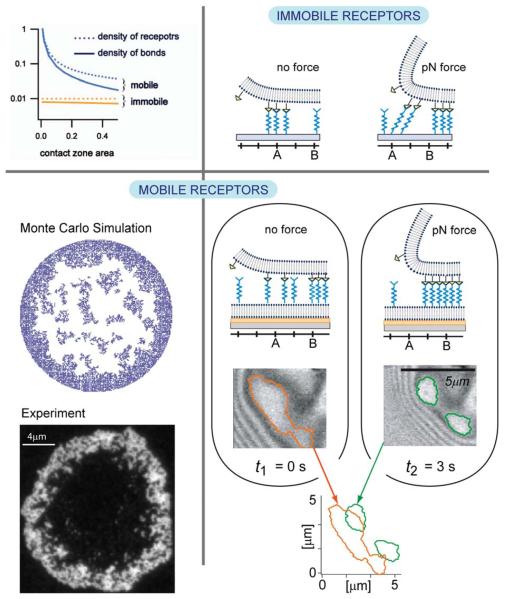
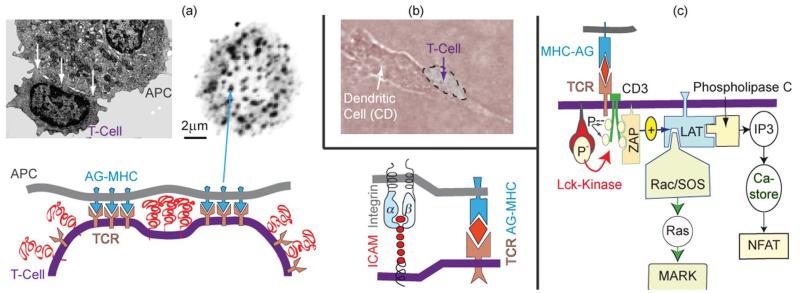
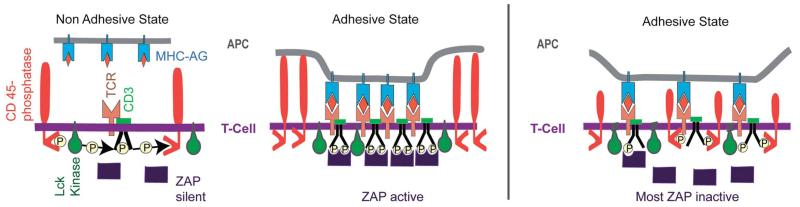
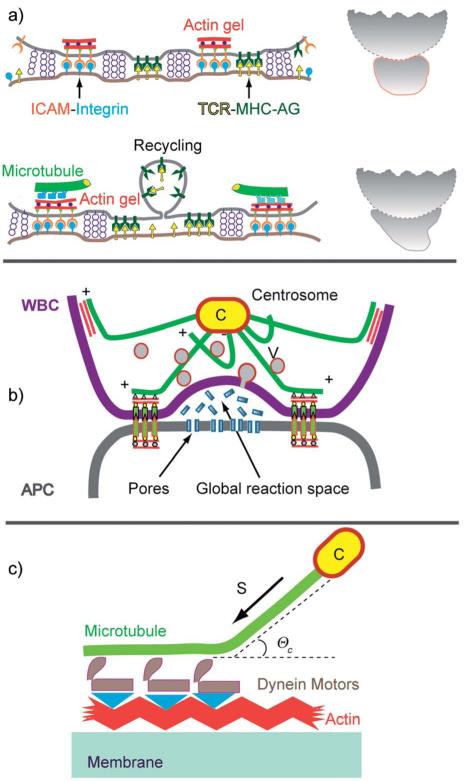
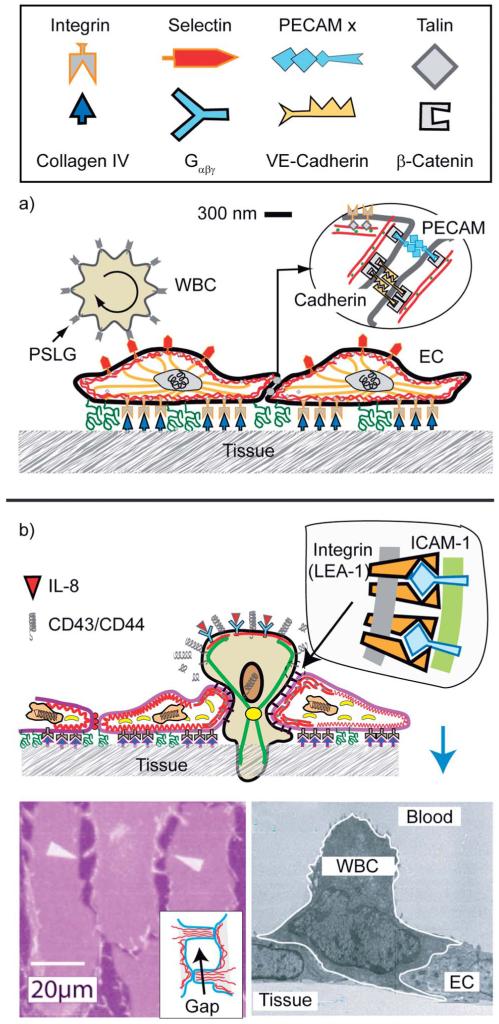
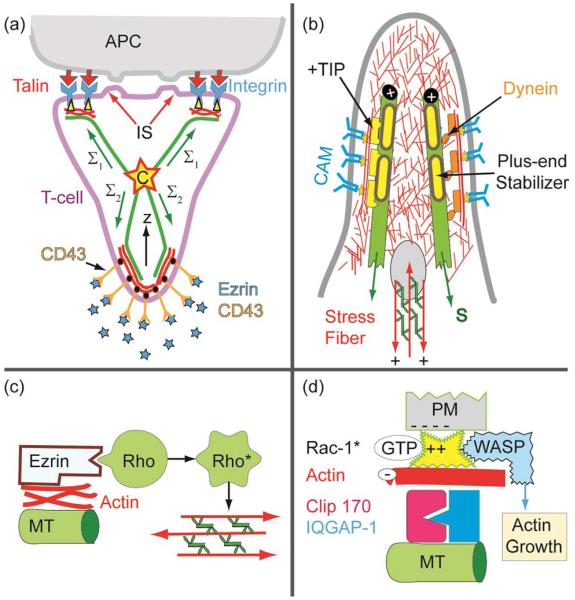
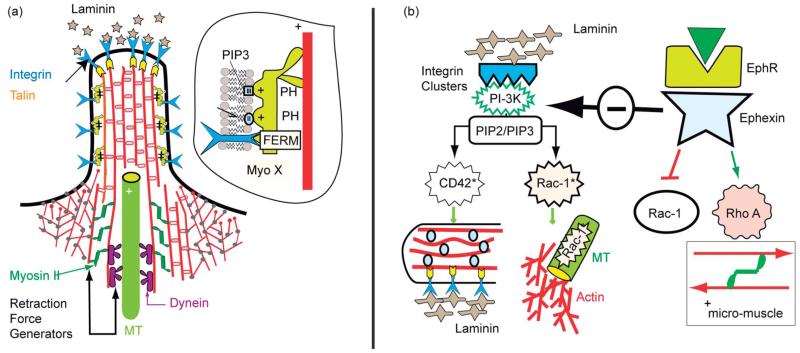
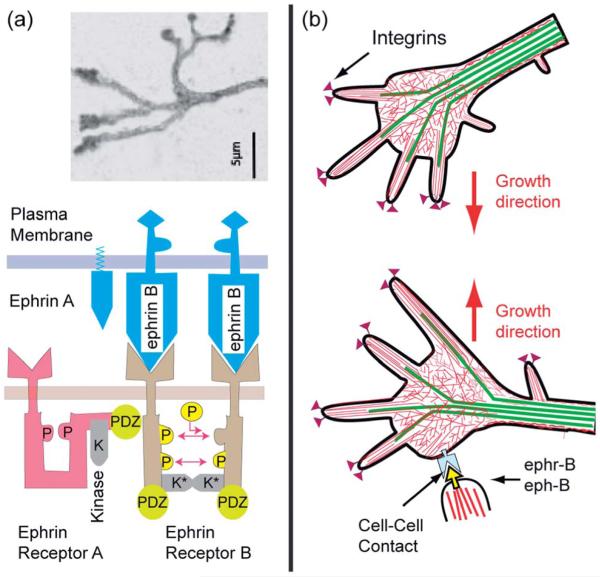
Cell adhesion is a paradigm of the ubiquitous interplay of cell signalling, modulation of material properties and biological functions of cells. It is controlled by competition of short range attractive forces, medium range repellant forces and the elastic stresses associated with local and global deformation of the composite cell envelopes. We review the basic physical rules governing the physics of cell adhesion learned by studying cell-mimetic systems and demonstrate the importance of these rules in the context of cellular systems. We review how adhesion induced micro-domains couple to the intracellular actin and microtubule networks allowing cells to generate strong forces with a minimum of attractive cell adhesion molecules (CAMs) and to manipulate other cells through filopodia over micrometer distances. The adhesion strength can be adapted to external force fluctuations within seconds by varying the density of attractive and repellant CAMs through exocytosis and endocytosis or protease-mediated dismantling of the CAM-cytoskeleton link. Adhesion domains form local end global biochemical reaction centres enabling the control of enzymes. Actin-microtubule crosstalk at adhesion foci facilitates the mechanical stabilization of polarized cell shapes. Axon growth in tissue is guided by attractive and repulsive clues controlled by antagonistic signalling pathways.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources