

Continuum approaches to understanding ion and peptide interactions with the membrane
- PMID: 24652510
- PMCID: PMC4096575
- DOI: 10.1007/s00232-014-9646-z
Continuum approaches to understanding ion and peptide interactions with the membrane
Abstract
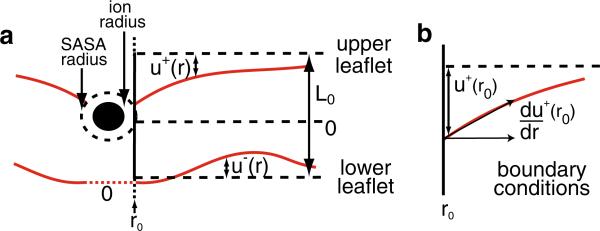
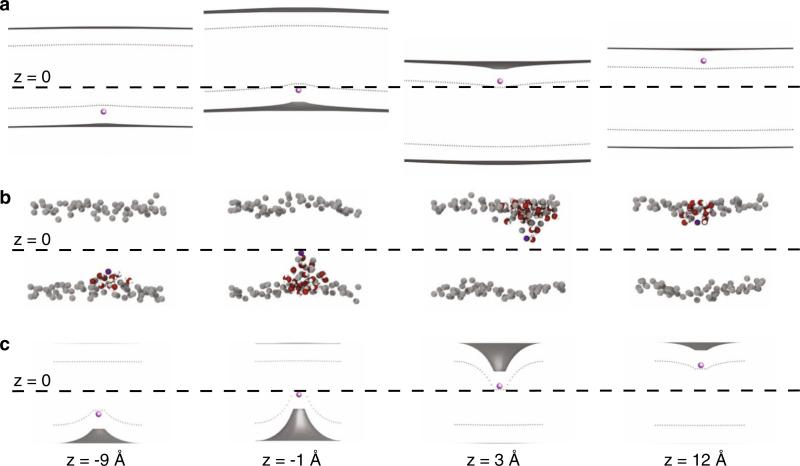
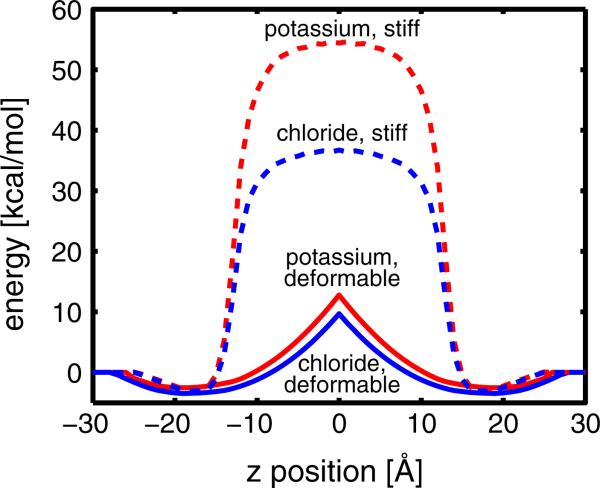
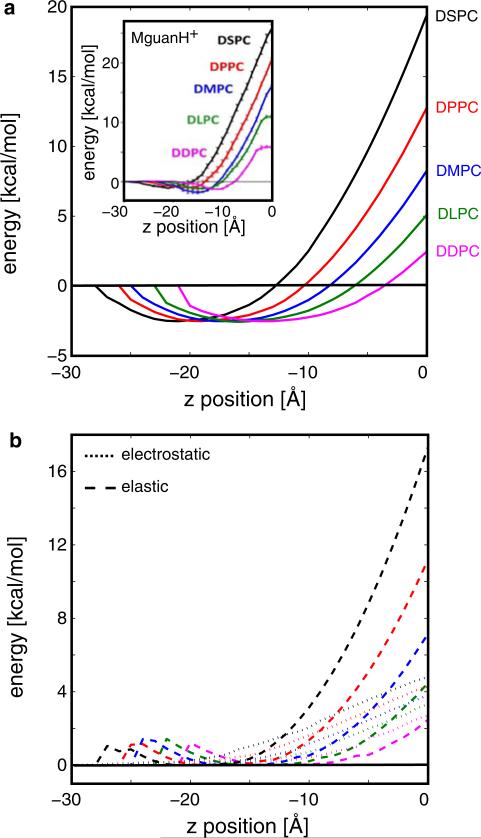
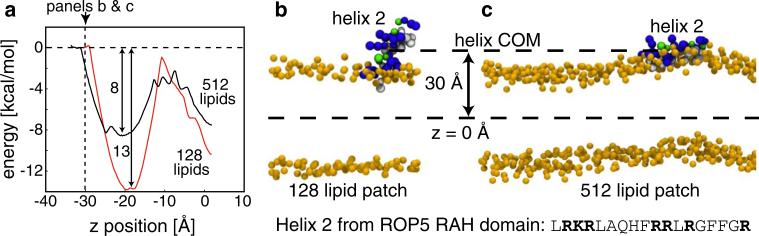
Experimental and computational studies have shown that cellular membranes deform to stabilize the inclusion of transmembrane (TM) proteins harboring charge. Recent analysis suggests that membrane bending helps to expose charged and polar residues to the aqueous environment and polar head groups. We previously used elasticity theory to identify membrane distortions that minimize the insertion of charged TM peptides into the membrane. Here, we extend our work by showing that it also provides a novel, computationally efficient method for exploring the energetics of ion and small peptide penetration into membranes. First, we show that the continuum method accurately reproduces energy profiles and membrane shapes generated from molecular simulations of bare ion permeation at a fraction of the computational cost. Next, we demonstrate that the dependence of the ion insertion energy on the membrane thickness arises primarily from the elastic properties of the membrane. Moreover, the continuum model readily provides a free energy decomposition into components not easily determined from molecular dynamics. Finally, we show that the energetics of membrane deformation strongly depend on membrane patch size both for ions and peptides. This dependence is particularly strong for peptides based on simulations of a known amphipathic, membrane binding peptide from the human pathogen Toxoplasma gondii. In total, we address shortcomings and advantages that arise from using a variety of computational methods in distinct biological contexts.
Figures
Similar articles
-
The importance of membrane defects-lessons from simulations.Acc Chem Res. 2014 Aug 19;47(8):2244-51. doi: 10.1021/ar4002729. Epub 2014 Jun 3. Acc Chem Res. 2014. PMID: 24892900
-
A continuum method for determining membrane protein insertion energies and the problem of charged residues.J Gen Physiol. 2008 Jun;131(6):563-73. doi: 10.1085/jgp.200809959. Epub 2008 May 12. J Gen Physiol. 2008. PMID: 18474636 Free PMC article.
-
Interactions of hydrophobic peptides with lipid bilayers: Monte Carlo simulations with M2delta.Biophys J. 2003 Dec;85(6):3431-44. doi: 10.1016/S0006-3495(03)74765-1. Biophys J. 2003. PMID: 14645040 Free PMC article.
-
Continuum descriptions of membranes and their interaction with proteins: Towards chemically accurate models.Biochim Biophys Acta. 2016 Jul;1858(7 Pt B):1619-34. doi: 10.1016/j.bbamem.2016.02.003. Epub 2016 Feb 4. Biochim Biophys Acta. 2016. PMID: 26853937 Free PMC article. Review.
-
Understanding and modelling the interactions of peptides with membranes: from partitioning to self-assembly.Curr Opin Struct Biol. 2020 Apr;61:160-166. doi: 10.1016/j.sbi.2019.12.021. Epub 2020 Jan 30. Curr Opin Struct Biol. 2020. PMID: 32006812 Review.
Cited by
-
Ion Permeation through a Phospholipid Membrane: Transition State, Path Splitting, and Calculation of Permeability.J Chem Theory Comput. 2019 Jan 8;15(1):720-730. doi: 10.1021/acs.jctc.8b00882. Epub 2018 Dec 7. J Chem Theory Comput. 2019. PMID: 30474968 Free PMC article.
-
Atomistic insight into lipid translocation by a TMEM16 scramblase.Proc Natl Acad Sci U S A. 2016 Dec 6;113(49):14049-14054. doi: 10.1073/pnas.1607574113. Epub 2016 Nov 21. Proc Natl Acad Sci U S A. 2016. PMID: 27872308 Free PMC article.
-
Molecular Basis of the Membrane Interaction of the β2e Subunit of Voltage-Gated Ca(2+) Channels.Biophys J. 2015 Sep 1;109(5):922-35. doi: 10.1016/j.bpj.2015.07.040. Biophys J. 2015. PMID: 26331250 Free PMC article.
-
Interactions of amino acid side-chain analogs within membrane environments.J Phys Chem B. 2015 Feb 19;119(7):2877-85. doi: 10.1021/jp511712u. Epub 2015 Feb 6. J Phys Chem B. 2015. PMID: 25621811 Free PMC article.
-
Modeling Membrane Curvature Generation due to Membrane⁻Protein Interactions.Biomolecules. 2018 Oct 23;8(4):120. doi: 10.3390/biom8040120. Biomolecules. 2018. PMID: 30360496 Free PMC article. Review.
References
-
- Bennett WD, Tieleman DP. Water defect and pore formation in atomistic and coarse-grained lipid membranes: pushing the limits of coarse graining. J Chem Theory Comput. 2011;7(9):2981–2988. - PubMed
-
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684. doi:10.1063/1.448118.
-
- Bond PJ, Wee CL, Sansom MS. Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry. 2008;47(43):11321–11331. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
