

Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons
- PMID: 24655393
- PMCID: PMC4116450
- DOI: 10.1111/acel.12216
Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons
Abstract
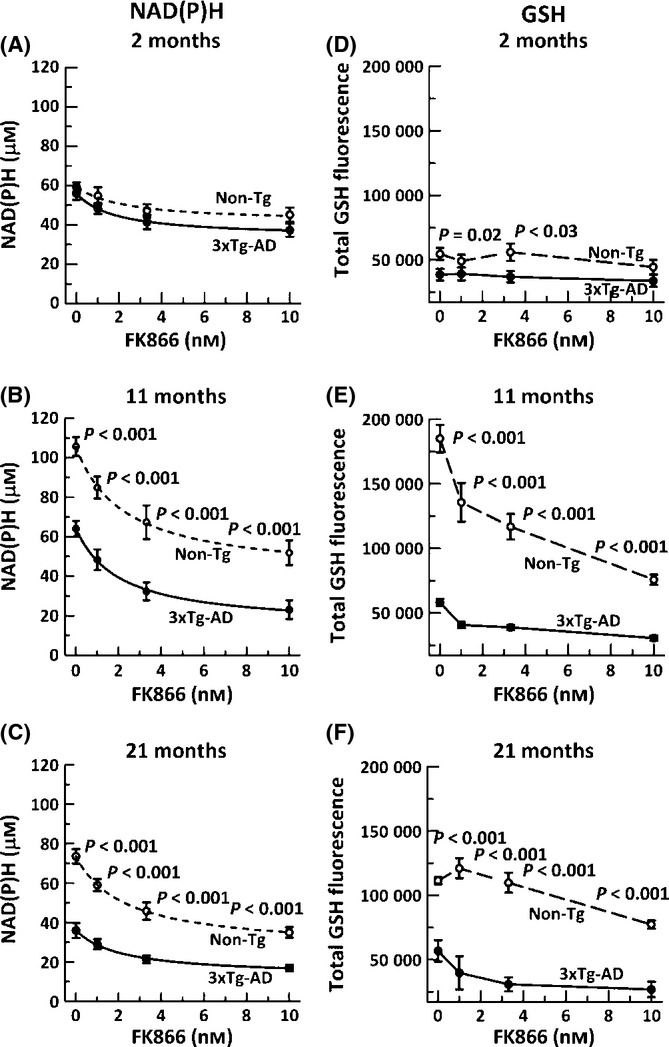
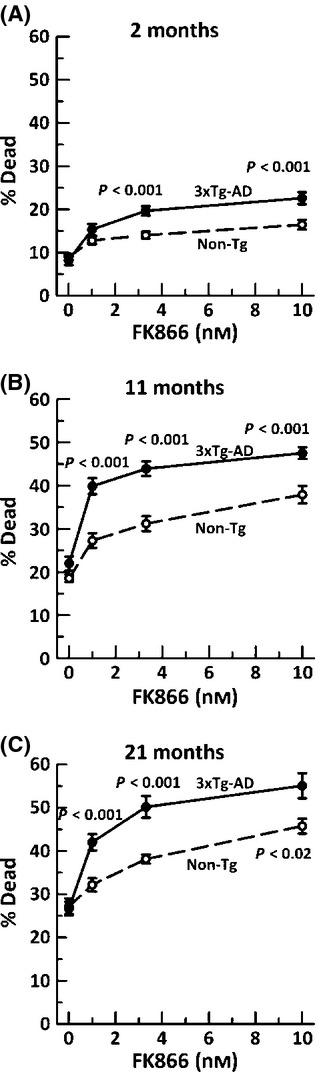
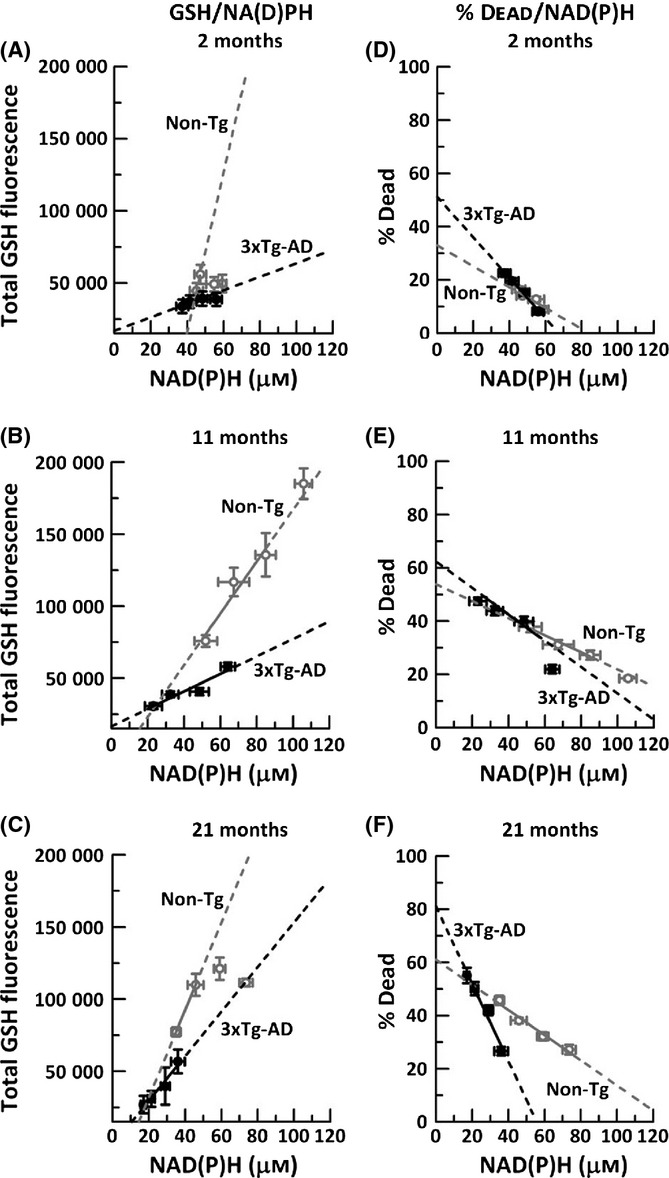
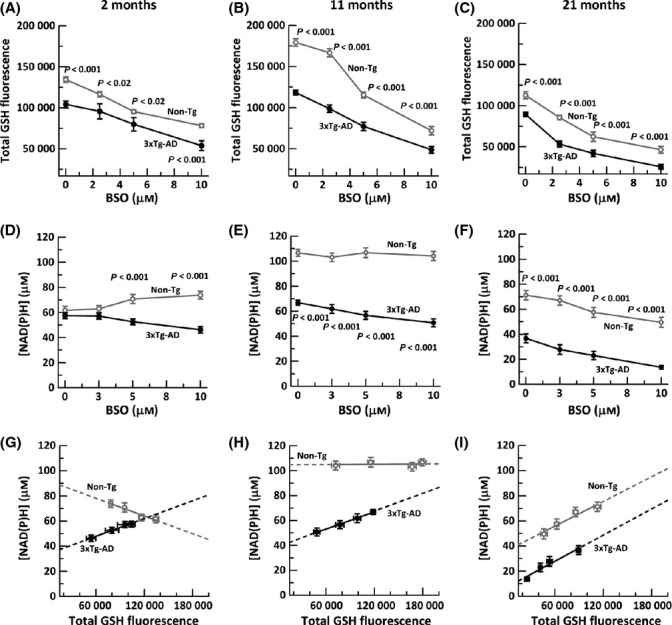
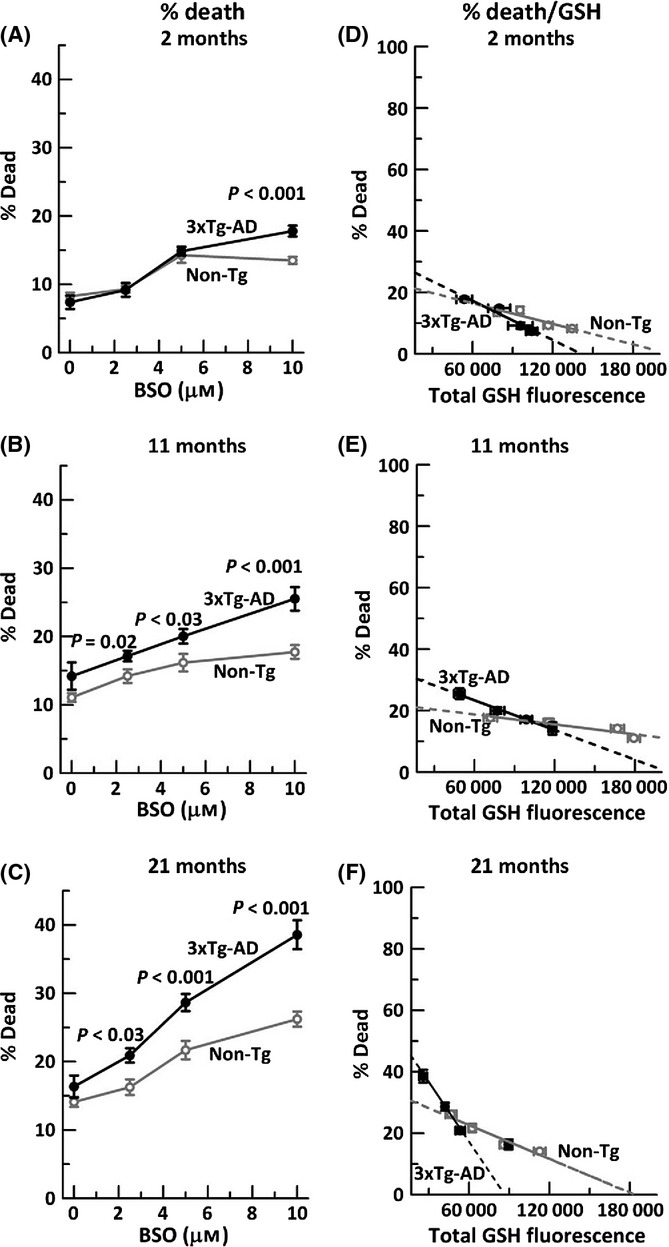
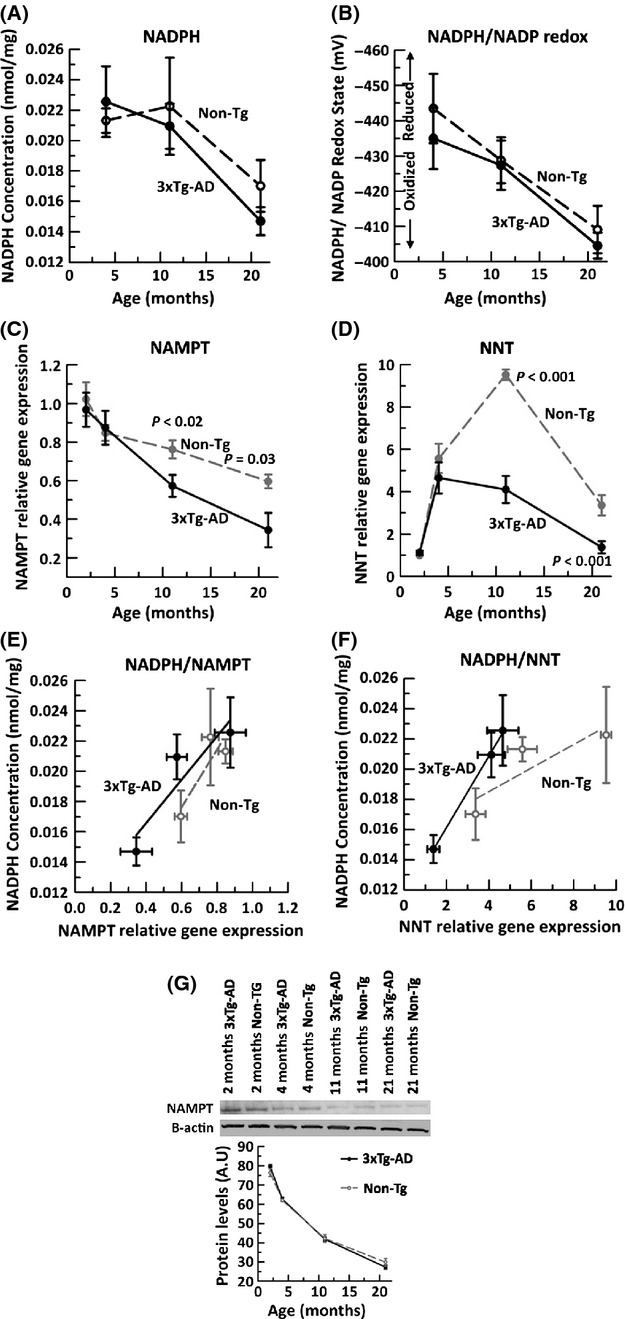
Aging, a major risk factor in Alzheimer's disease (AD), is associated with an oxidative redox shift, decreased redox buffer protection, and increased free radical reactive oxygen species (ROS) generation, probably linked to mitochondrial dysfunction. While NADH is the ultimate electron donor for many redox reactions, including oxidative phosphorylation, glutathione (GSH) is the major ROS detoxifying redox buffer in the cell. Here, we explored the relative importance of NADH and GSH to neurodegeneration in aging and AD neurons from nontransgenic and 3xTg-AD mice by inhibiting their synthesis to determine whether NADH can compensate for the GSH loss to maintain redox balance. Neurons stressed by either depleting NAD(P)H or GSH indicated that NADH redox control is upstream of GSH levels. Further, although depletion of NAD(P)H or GSH correlated linearly with neuron death, compared with GSH depletion, higher neurodegeneration was observed when NAD(P)H was extrapolated to zero, especially in old age, and in the 3xTg-AD neurons. We also observed an age-dependent loss of gene expression of key redox-dependent biosynthetic enzymes, NAMPT (nicotinamide phosphoribosyltransferase), and NNT (nicotinamide nucleotide transhydrogenase). Moreover, age-related correlations between brain NNT or NAMPT gene expression and NADPH levels suggest that these genes contribute to the age-related declines in NAD(P)H. Our data indicate that in aging and more so in AD-like neurons, NAD(P)H redox control is upstream of GSH and an oxidative redox shift that promotes neurodegeneration. Thus, NAD(P)H generation may be a more efficacious therapeutic target upstream of GSH and ROS.
Keywords: 3xTg-AD; NAD(P)H; aging; glutathione; neurodegeneration; redox.
© 2014 The Authors. Aging Cell published by the Anatomical Society and John Wiley & Sons Ltd.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
