

PTEN function: the long and the short of it
- PMID: 24656806
- PMCID: PMC4043120
- DOI: 10.1016/j.tibs.2014.02.006
PTEN function: the long and the short of it
Abstract
Phosphatase and tensin homolog deleted on chromosome ten (PTEN) is a phosphatase that is frequently altered in cancer. PTEN has phosphatase-dependent and -independent roles, and genetic alterations in PTEN lead to deregulation of protein synthesis, the cell cycle, migration, growth, DNA repair, and survival signaling. PTEN localization, stability, conformation, and phosphatase activity are controlled by an array of protein-protein interactions and post-translational modifications. Thus, PTEN-interacting and -modifying proteins have profound effects on the tumor suppressive functions of PTEN. Moreover, recent studies identified mechanisms by which PTEN can exit cells, via either exosomal export or secretion, and act on neighboring cells. This review focuses on modes of PTEN protein regulation and ways in which perturbations in this regulation may lead to disease.
Keywords: PI3K signaling; PTEN; PTEN-Long.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
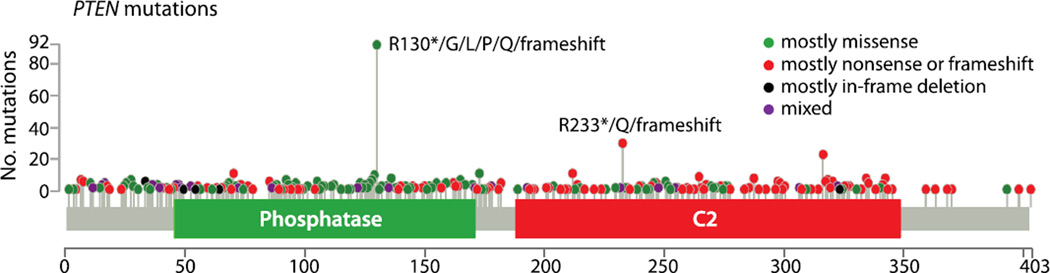
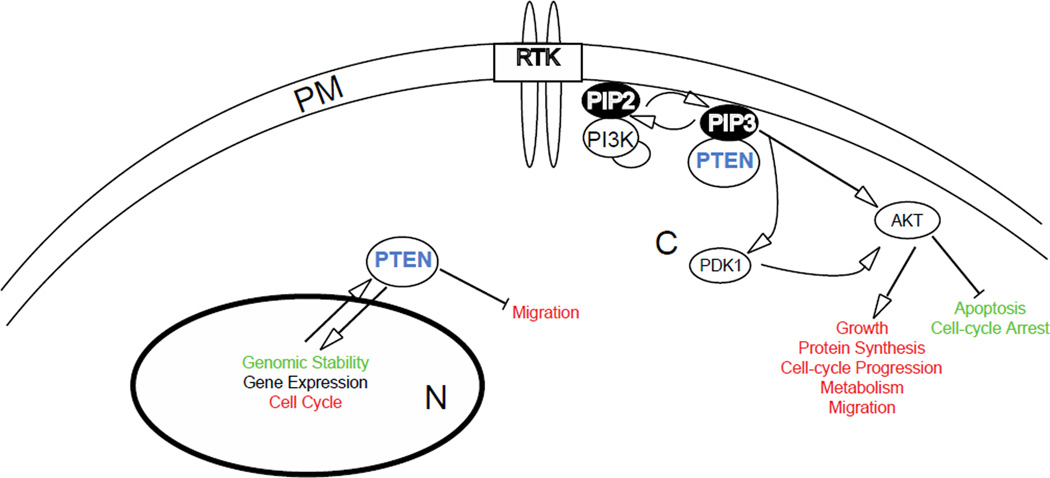
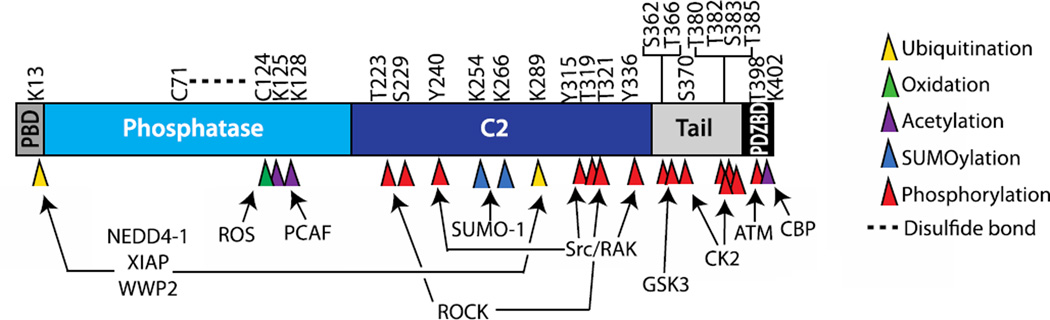
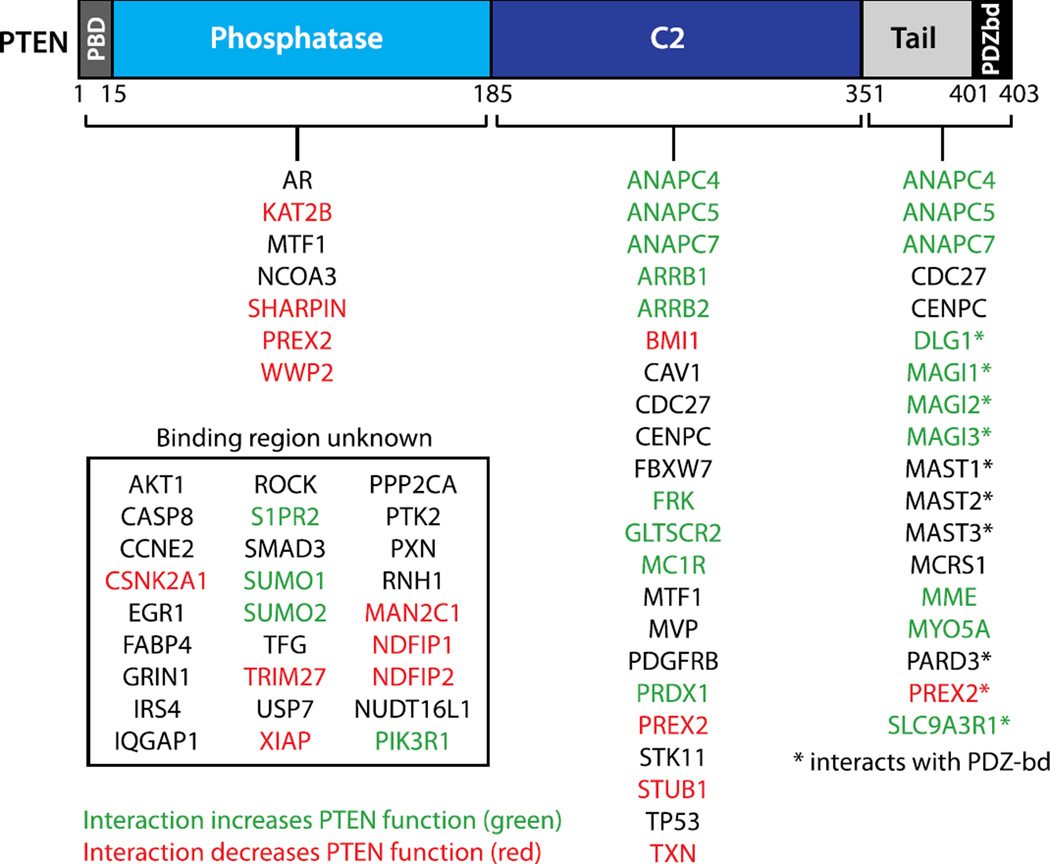
References
-
- Li J, et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science. 1997;275:1943–1947. - PubMed
-
- Steck PA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature genetics. 1997;15:356–362. - PubMed
-
- Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer research. 1997;57:2124–2129. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
