

Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2
- PMID: 24657168
- PMCID: PMC4011022
- DOI: 10.1016/j.molcel.2014.02.020
Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2
Erratum in
-
Inositol Pyrophosphates Mediate the DNA-PK/ATM-p53 Cell Death Pathway by Regulating CK2 Phosphorylation of Tti1/Tel2.Mol Cell. 2020 Aug 20;79(4):702. doi: 10.1016/j.molcel.2020.07.021. Mol Cell. 2020. PMID: 32822581 Free PMC article. No abstract available.
Abstract
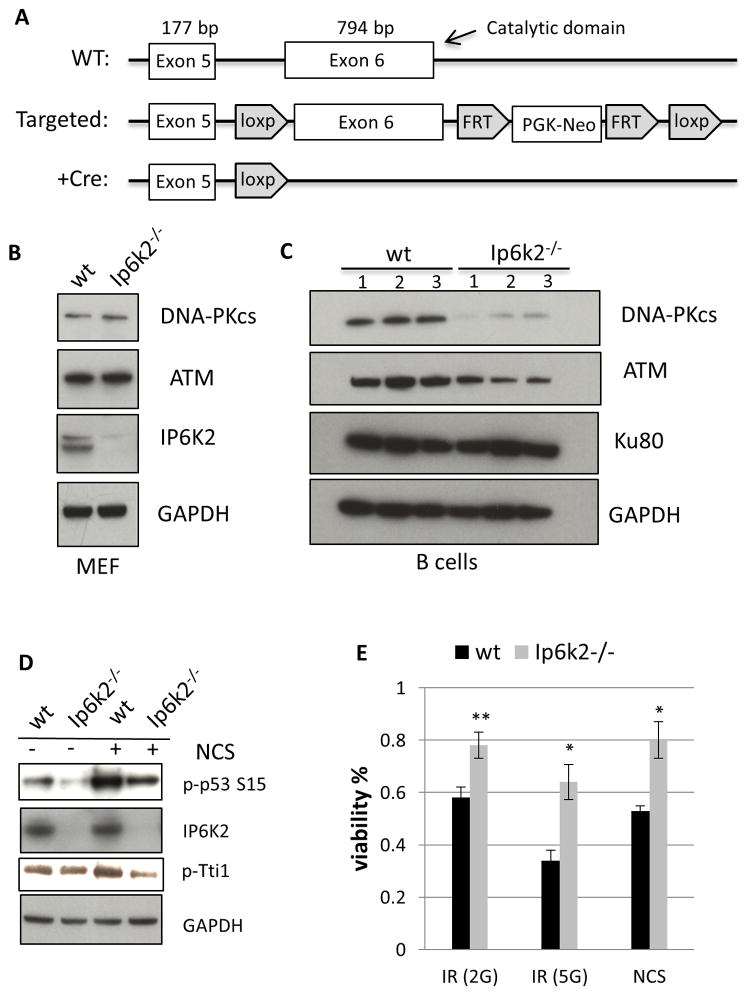
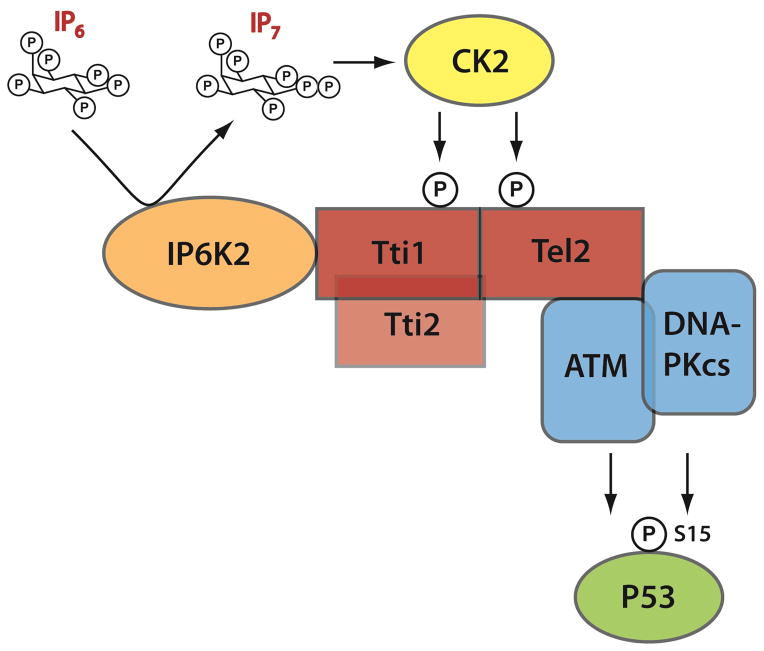
The apoptotic actions of p53 require its phosphorylation by a family of phosphoinositide-3-kinase-related-kinases (PIKKs), which include DNA-PKcs and ATM. These kinases are stabilized by the TTT (Tel2, Tti1, Tti2) cochaperone family, whose actions are mediated by CK2 phosphorylation. The inositol pyrophosphates, such as 5-diphosphoinositol pentakisphosphate (IP7), are generated by a family of inositol hexakisphosphate kinases (IP6Ks), of which IP6K2 has been implicated in p53-associated cell death. In the present study we report an apoptotic signaling cascade linking CK2, TTT, the PIKKs, and p53. We demonstrate that IP7, formed by IP6K2, binds CK2 to enhance its phosphorylation of the TTT complex, thereby stabilizing DNA-PKcs and ATM. This process stimulates p53 phosphorylation at serine 15 to activate the cell death program in human cancer cells and in murine B cells.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
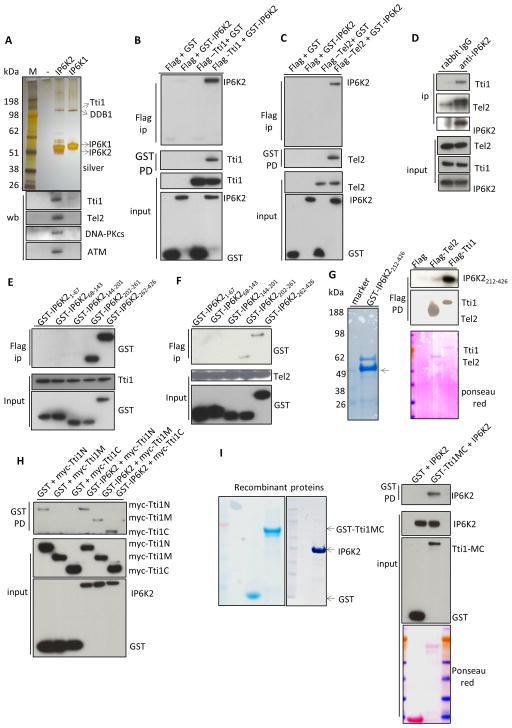
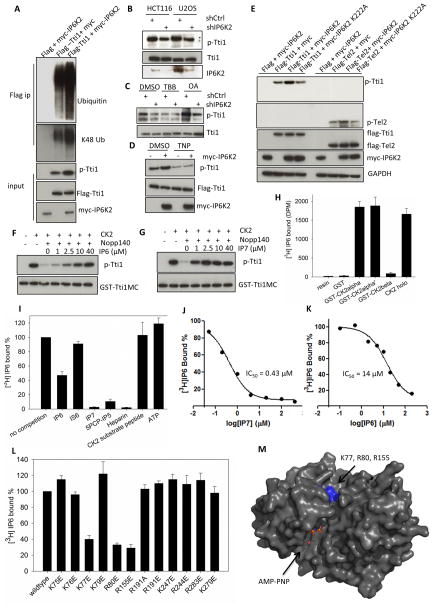
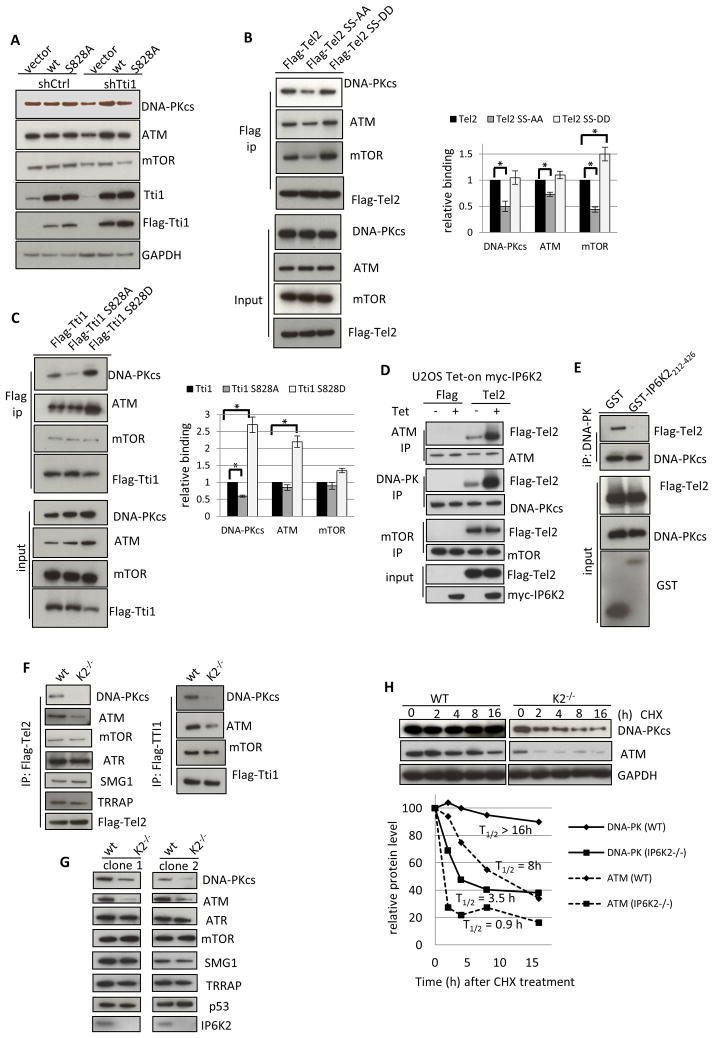
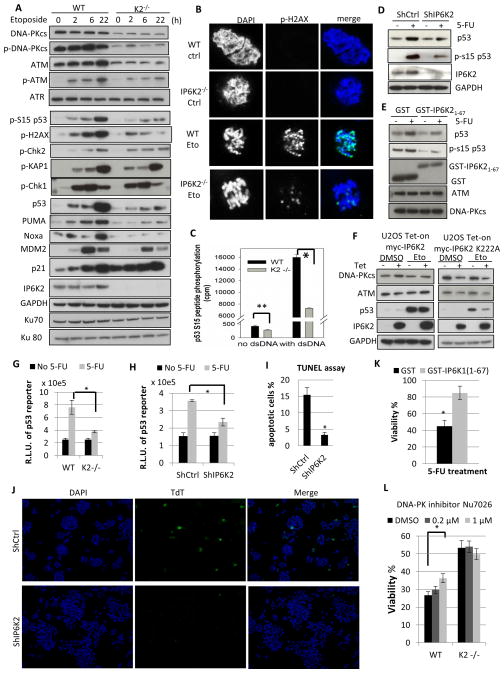
References
-
- Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3:883–887. - PubMed
-
- Achari Y, Lees-Miller SP. Detection of DNA-dependent protein kinase in extracts from human and rodent cells. Methods Mol Biol. 2000;99:85–97. - PubMed
-
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
