

Endothelial dysfunction in adipose triglyceride lipase deficiency
- PMID: 24657704
- PMCID: PMC4000266
- DOI: 10.1016/j.bbalip.2014.03.005
Endothelial dysfunction in adipose triglyceride lipase deficiency
Abstract
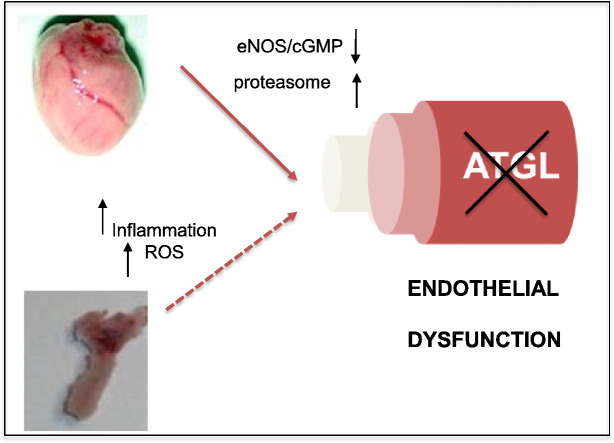
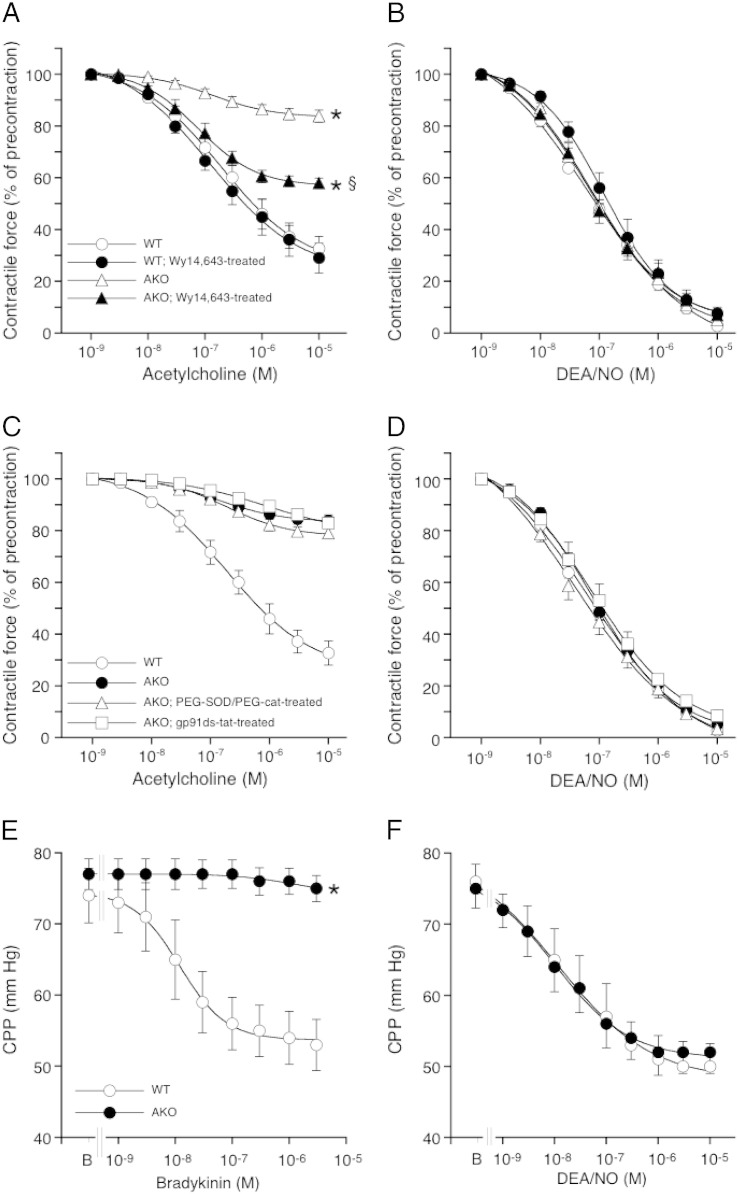
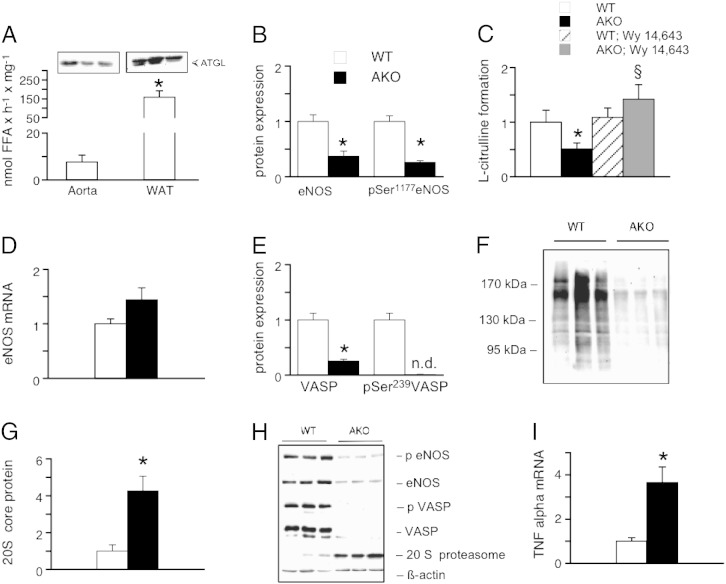
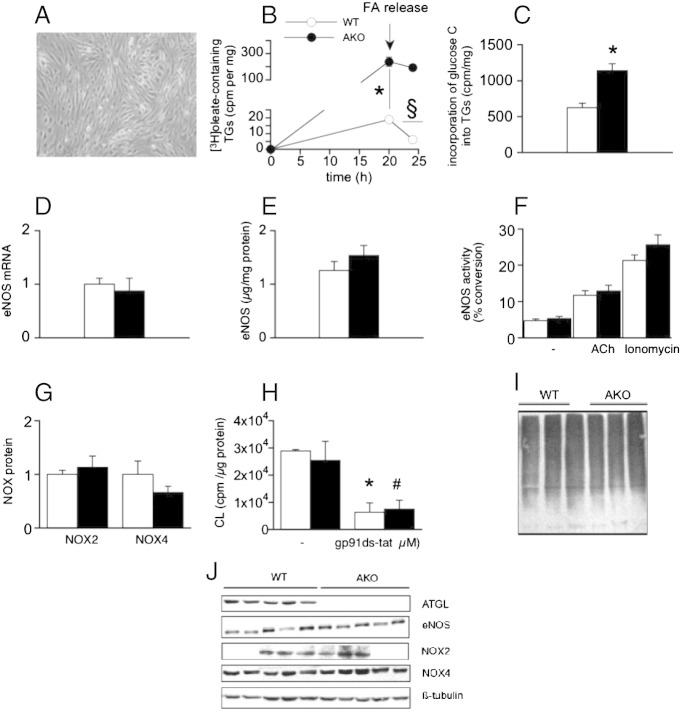
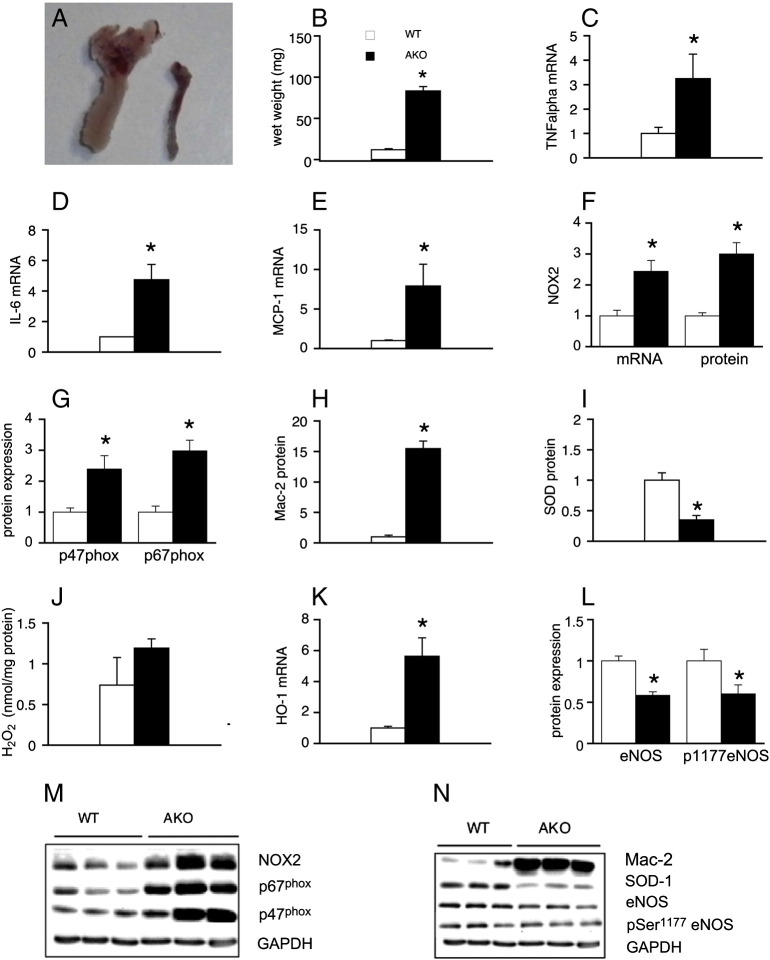
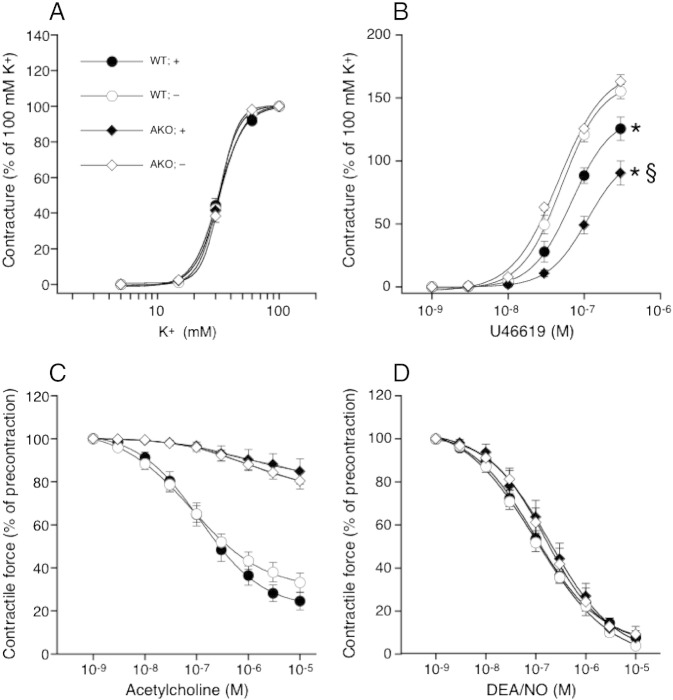
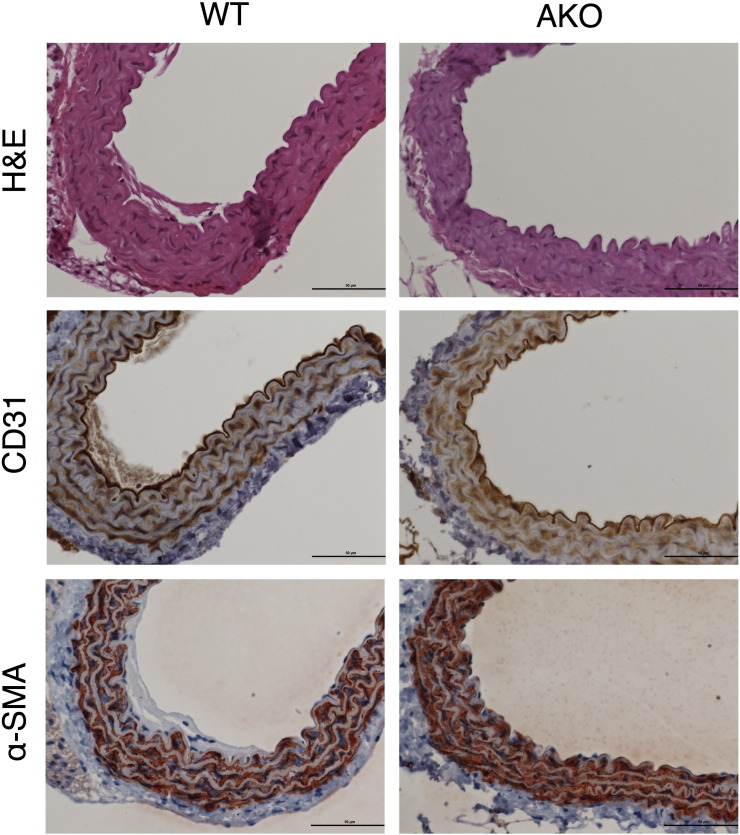
Systemic knockout of adipose triglyceride lipase (ATGL), the pivotal enzyme of triglyceride lipolysis, results in a murine phenotype that is characterized by progredient cardiac steatosis and severe heart failure. Since cardiac and vascular dysfunction have been closely related in numerous studies we investigated endothelium-dependent and -independent vessel function of ATGL knockout mice. Aortic relaxation studies and Langendorff perfusion experiments of isolated hearts showed that ATGL knockout mice suffer from pronounced micro- and macrovascular endothelial dysfunction. Experiments with agonists directly targeting vascular smooth muscle cells revealed the functional integrity of the smooth muscle cell layer. Loss of vascular reactivity was restored ~50% upon treatment of ATGL knockout mice with the PPARα agonist Wy14,643, indicating that this phenomenon is partly a consequence of impaired cardiac contractility. Biochemical analysis revealed that aortic endothelial NO synthase expression and activity were significantly reduced in ATGL deficiency. Enzyme activity was fully restored in ATGL mice treated with the PPARα agonist. Biochemical analysis of perivascular adipose tissue demonstrated that ATGL knockout mice suffer from perivascular inflammatory oxidative stress which occurs independent of cardiac dysfunction and might contribute to vascular defects. Our results reveal a hitherto unrecognized link between disturbed lipid metabolism, obesity and cardiovascular disease.
Keywords: Adipose triglyceride lipase; Endothelial NO synthase; Endothelial dysfunction; Perivascular inflammation; Vascular proteasome.
Copyright © 2014 The Authors. Published by Elsevier B.V. All rights reserved.
Figures

Similar articles
-
Role of the ubiquitin-proteasome system in cardiac dysfunction of adipose triglyceride lipase-deficient mice.J Mol Cell Cardiol. 2014 Dec;77:11-9. doi: 10.1016/j.yjmcc.2014.09.028. Epub 2014 Oct 5. J Mol Cell Cardiol. 2014. PMID: 25285770 Free PMC article.
-
Cardiac oxidative stress in a mouse model of neutral lipid storage disease.Biochim Biophys Acta. 2013 Nov;1831(11):1600-8. doi: 10.1016/j.bbalip.2013.07.004. Epub 2013 Jul 15. Biochim Biophys Acta. 2013. PMID: 23867907 Free PMC article.
-
Cardiac dysfunction in adipose triglyceride lipase deficiency: treatment with a PPARα agonist.Br J Pharmacol. 2012 Jan;165(2):380-9. doi: 10.1111/j.1476-5381.2011.01490.x. Br J Pharmacol. 2012. PMID: 21585347 Free PMC article.
-
The role of adipose triglyceride lipase in lipid and glucose homeostasis: lessons from transgenic mice.Lipids Health Dis. 2019 Nov 22;18(1):204. doi: 10.1186/s12944-019-1151-z. Lipids Health Dis. 2019. PMID: 31757217 Free PMC article. Review.
-
[Adipose triglyceride lipase regulates adipocyte lipolysis].Sheng Li Ke Xue Jin Zhan. 2008 Jan;39(1):10-4. Sheng Li Ke Xue Jin Zhan. 2008. PMID: 18357681 Review. Chinese.
Cited by
-
The role of perivascular adipose tissue in vasoconstriction, arterial stiffness, and aneurysm.Horm Mol Biol Clin Investig. 2015 Feb;21(2):137-47. doi: 10.1515/hmbci-2014-0048. Horm Mol Biol Clin Investig. 2015. PMID: 25719334 Free PMC article. Review.
-
Vascular ATGL-dependent lipolysis and the activation of cPLA2-PGI2 pathway protect against postprandial endothelial dysfunction.Cell Mol Life Sci. 2024 Mar 12;81(1):125. doi: 10.1007/s00018-024-05167-6. Cell Mol Life Sci. 2024. PMID: 38467757 Free PMC article.
-
Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis.J Leukoc Biol. 2015 Nov;98(5):837-50. doi: 10.1189/jlb.3A0515-206R. Epub 2015 Jun 24. J Leukoc Biol. 2015. PMID: 26109679 Free PMC article.
-
Lipid droplets in the endothelium: The missing link between metabolic syndrome and cardiovascular disease?J Clin Invest. 2024 Feb 15;134(4):e176347. doi: 10.1172/JCI176347. J Clin Invest. 2024. PMID: 38357921 Free PMC article.
-
Vascular lipid droplets formed in response to TNF, hypoxia, or OA: biochemical composition and prostacyclin generation.J Lipid Res. 2023 May;64(5):100355. doi: 10.1016/j.jlr.2023.100355. Epub 2023 Mar 17. J Lipid Res. 2023. PMID: 36934842 Free PMC article.
References
-
- Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., Kratky D., Wagner E.F., Klingenspor M., Hoefler G., Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. - PubMed
-
- Haemmerle G., Moustafa T., Woelkart G., Buttner S., Schmidt A., van de Weijer T., Hesselink M., Jaeger D., Kienesberger P.C., Zierler K., Schreiber R., Eichmann T., Kolb D., Kotzbeck P., Schweiger M., Kumari M., Eder S., Schoiswohl G., Wongsiriroj N., Pollak N.M., Radner F.P., Preiss-Landl K., Kolbe T., Rulicke T., Pieske B., Trauner M., Lass A., Zimmermann R., Hoefler G., Cinti S., Kershaw E.E., Schrauwen P., Madeo F., Mayer B., Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011;17:1076–1085. - PMC - PubMed
-
- Bauersachs J., Widder J.D. Endothelial dysfunction in heart failure. Pharmacol. Rep. 2008;60:119–126. - PubMed
-
- Fischer D., Rossa S., Landmesser U., Spiekermann S., Engberding N., Hornig B., Drexler H. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur. Heart J. 2005;26:65–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
