

Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury
- PMID: 24659784
- PMCID: PMC4036326
- DOI: 10.1074/jbc.M113.530311
Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury
Abstract
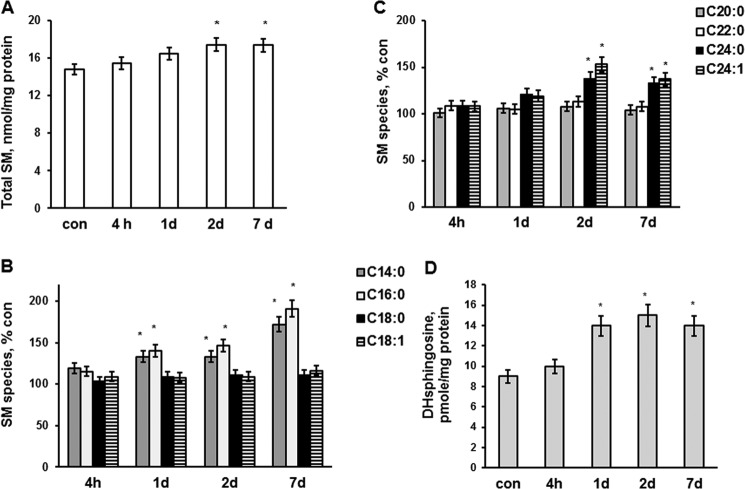
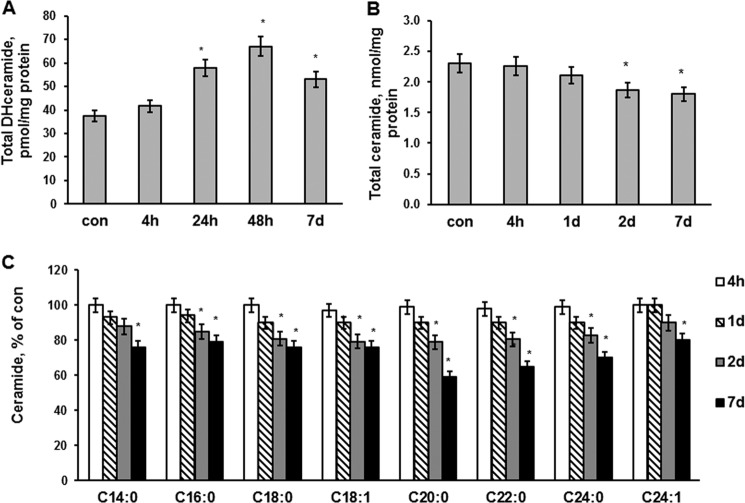
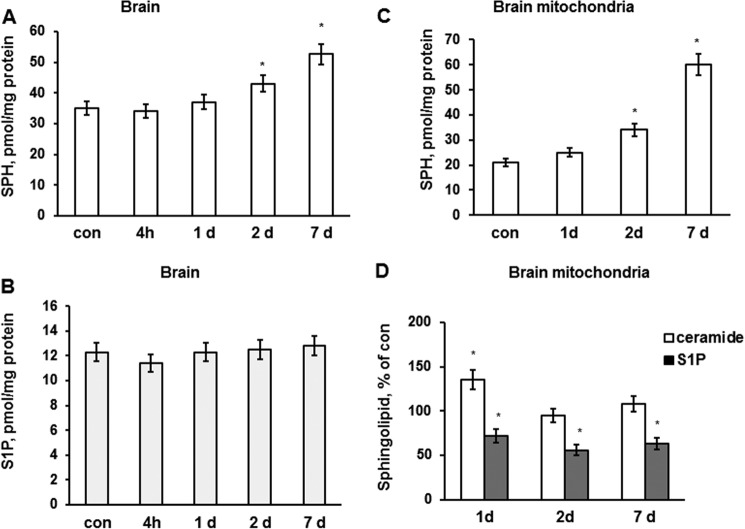
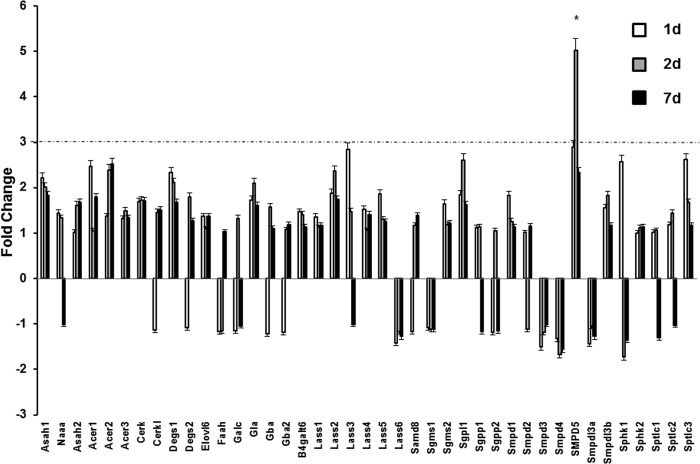
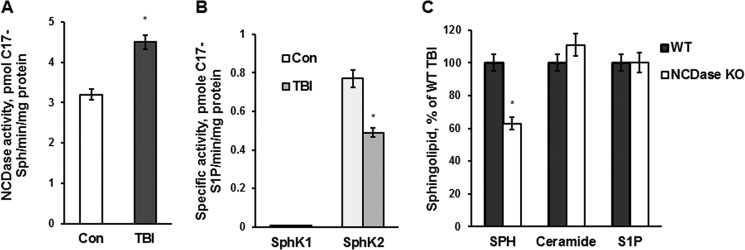
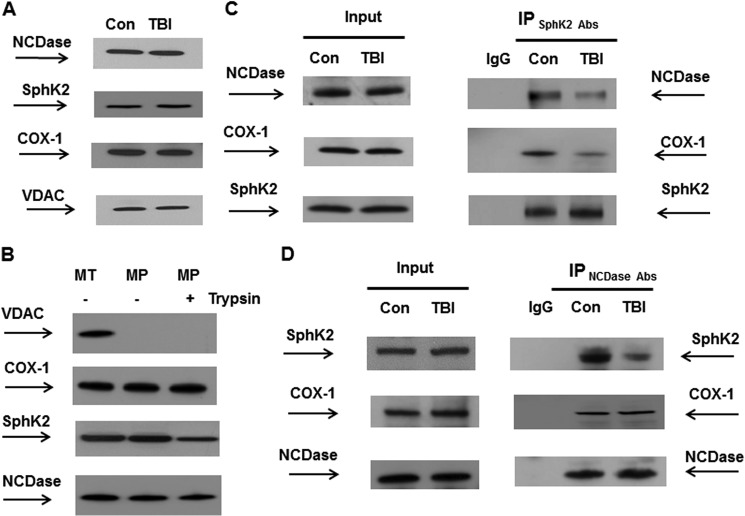
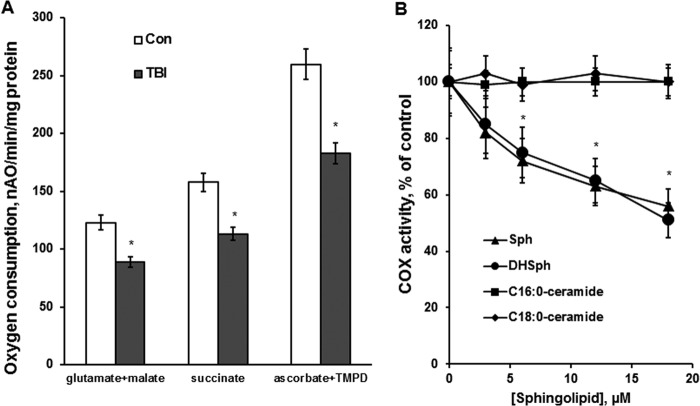
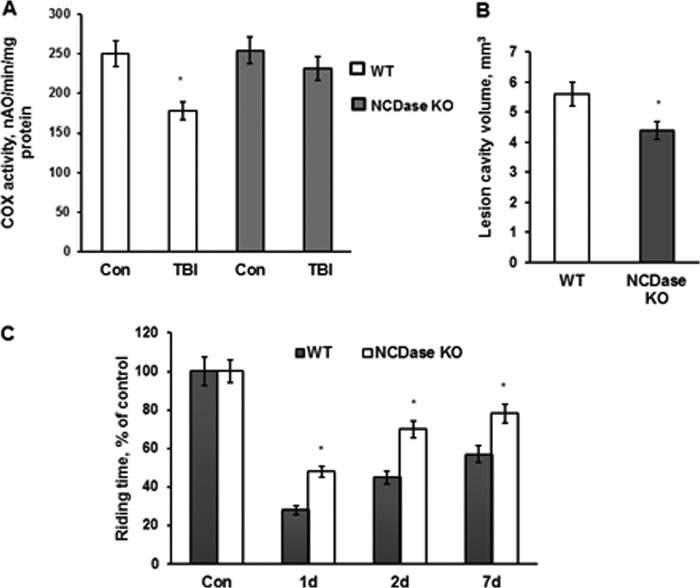
In addition to immediate brain damage, traumatic brain injury (TBI) initiates a cascade of pathophysiological events producing secondary injury. The biochemical and cellular mechanisms that comprise secondary injury are not entirely understood. Herein, we report a substantial deregulation of cerebral sphingolipid metabolism in a mouse model of TBI. Sphingolipid profile analysis demonstrated increases in sphingomyelin species and sphingosine concurrently with up-regulation of intermediates of de novo sphingolipid biosynthesis in the brain. Investigation of intracellular sites of sphingosine accumulation revealed an elevation of sphingosine in mitochondria due to the activation of neutral ceramidase (NCDase) and the reduced activity of sphingosine kinase 2 (SphK2). The lack of change in gene expression suggested that post-translational mechanisms are responsible for the shift in the activities of both enzymes. Immunoprecipitation studies revealed that SphK2 is complexed with NCDase and cytochrome oxidase (COX) subunit 1 in mitochondria and that brain injury hindered SphK2 association with the complex. Functional studies showed that sphingosine accumulation resulted in a decreased activity of COX, a rate-limiting enzyme of the mitochondrial electron transport chain. Knocking down NCDase reduced sphingosine accumulation in mitochondria and preserved COX activity after the brain injury. Also, NCDase knockdown improved brain function recovery and lessened brain contusion volume after trauma. These studies highlight a novel mechanism of secondary TBI involving a disturbance of sphingolipid-metabolizing enzymes in mitochondria and suggest a critical role for mitochondrial sphingosine in promoting brain injury after trauma.
Keywords: Brain; Cell Death; Mitochondria; Respiratory Chain; Sphingolipid.
Figures
Similar articles
-
Traumatic brain injury-induced changes in gene expression and functional activity of mitochondrial cytochrome C oxidase.J Neurotrauma. 2001 Oct;18(10):993-1009. doi: 10.1089/08977150152693692. J Neurotrauma. 2001. PMID: 11686499
-
Novel pathway of ceramide production in mitochondria: thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA.J Biol Chem. 2011 Jul 15;286(28):25352-62. doi: 10.1074/jbc.M110.214866. Epub 2011 May 25. J Biol Chem. 2011. PMID: 21613224 Free PMC article.
-
Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson's disease and in MPP+ -treated MN9D cells in vitro.Neuroscience. 2015 Apr 2;290:636-48. doi: 10.1016/j.neuroscience.2015.01.032. Epub 2015 Jan 28. Neuroscience. 2015. PMID: 25637806
-
Colon Cancer and Perturbations of the Sphingolipid Metabolism.Int J Mol Sci. 2019 Nov 30;20(23):6051. doi: 10.3390/ijms20236051. Int J Mol Sci. 2019. PMID: 31801289 Free PMC article. Review.
-
Ceramidases, roles in sphingolipid metabolism and in health and disease.Adv Biol Regul. 2017 Jan;63:122-131. doi: 10.1016/j.jbior.2016.10.002. Epub 2016 Oct 11. Adv Biol Regul. 2017. PMID: 27771292 Free PMC article. Review.
Cited by
-
SIRT3 Deacetylates Ceramide Synthases: IMPLICATIONS FOR MITOCHONDRIAL DYSFUNCTION AND BRAIN INJURY.J Biol Chem. 2016 Jan 22;291(4):1957-1973. doi: 10.1074/jbc.M115.668228. Epub 2015 Nov 30. J Biol Chem. 2016. PMID: 26620563 Free PMC article.
-
Integrative Analysis of Cytokine and Lipidomics Datasets Following Mild Traumatic Brain Injury in Rats.Metabolites. 2024 Feb 21;14(3):133. doi: 10.3390/metabo14030133. Metabolites. 2024. PMID: 38535293 Free PMC article.
-
Sphingosine kinase 1 downregulation is required for adaptation to serine deprivation.FASEB J. 2021 Feb;35(2):e21284. doi: 10.1096/fj.202001814RR. FASEB J. 2021. PMID: 33484475 Free PMC article.
-
Sphingolipids and mitochondrial apoptosis.J Bioenerg Biomembr. 2016 Apr;48(2):153-68. doi: 10.1007/s10863-015-9602-3. J Bioenerg Biomembr. 2016. PMID: 25620271 Free PMC article. Review.
-
Sphingolipid changes in mouse brain and plasma after mild traumatic brain injury at the acute phases.Lipids Health Dis. 2024 Jun 27;23(1):200. doi: 10.1186/s12944-024-02186-x. Lipids Health Dis. 2024. PMID: 38937745 Free PMC article.
References
-
- Spaethling J. M., Geddes-Klein D. M., Miller W. J., von Reyn C. R., Singh P., Mesfin M., Bernstein S. J., Meaney D. F. (2007) Linking impact to cellular and molecular sequelae of CNS injury: modeling in vivo complexity with in vitro simplicity. Prog. Brain Res. 161, 27–39 - PubMed
-
- Sullivan P. G., Keller J. N., Mattson M. P., Scheff S. W. (1998) Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J. Neurotrauma 15, 789–798 - PubMed
-
- Singh I. N., Sullivan P. G., Hall E. D. (2007) Peroxynitrite-mediated oxidative damage to brain mitochondria: protective effects of peroxynitrite scavengers. J. Neurosci. Res. 85, 2216–2223 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
