

Supersize me: how whole-genome sequencing and big data are transforming epidemiology
- PMID: 24661923
- PMCID: PMC7125769
- DOI: 10.1016/j.tim.2014.02.011
Supersize me: how whole-genome sequencing and big data are transforming epidemiology
Abstract
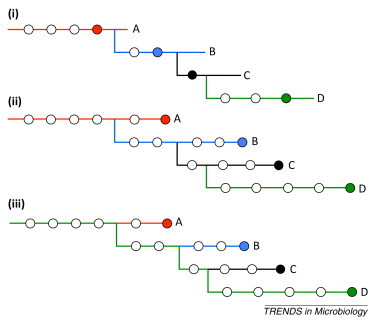
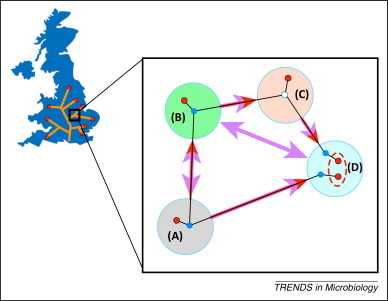
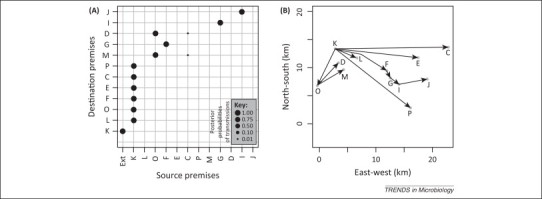
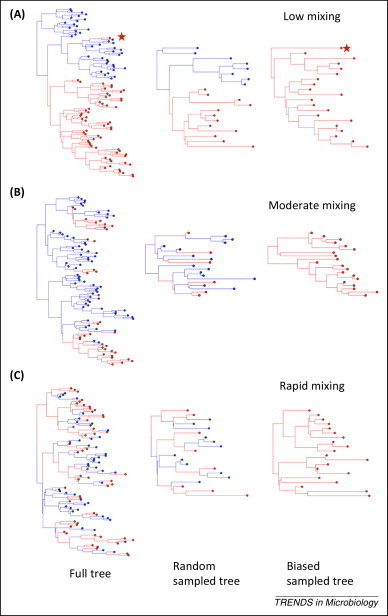
In epidemiology, the identification of 'who infected whom' allows us to quantify key characteristics such as incubation periods, heterogeneity in transmission rates, duration of infectiousness, and the existence of high-risk groups. Although invaluable, the existence of many plausible infection pathways makes this difficult, and epidemiological contact tracing either uncertain, logistically prohibitive, or both. The recent advent of next-generation sequencing technology allows the identification of traceable differences in the pathogen genome that are transforming our ability to understand high-resolution disease transmission, sometimes even down to the host-to-host scale. We review recent examples of the use of pathogen whole-genome sequencing for the purpose of forensic tracing of transmission pathways, focusing on the particular problems where evolutionary dynamics must be supplemented by epidemiological information on the most likely timing of events as well as possible transmission pathways. We also discuss potential pitfalls in the over-interpretation of these data, and highlight the manner in which a confluence of this technology with sophisticated mathematical and statistical approaches has the potential to produce a paradigm shift in our understanding of infectious disease transmission and control.
Keywords: Bayesian inference; Forensic epidemiology; Mathematical modeling; Pathogen evolution; Who-infected-whom?.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
