

Fast and accurate multivariate Gaussian modeling of protein families: predicting residue contacts and protein-interaction partners
- PMID: 24663061
- PMCID: PMC3963956
- DOI: 10.1371/journal.pone.0092721
Fast and accurate multivariate Gaussian modeling of protein families: predicting residue contacts and protein-interaction partners
Abstract
In the course of evolution, proteins show a remarkable conservation of their three-dimensional structure and their biological function, leading to strong evolutionary constraints on the sequence variability between homologous proteins. Our method aims at extracting such constraints from rapidly accumulating sequence data, and thereby at inferring protein structure and function from sequence information alone. Recently, global statistical inference methods (e.g. direct-coupling analysis, sparse inverse covariance estimation) have achieved a breakthrough towards this aim, and their predictions have been successfully implemented into tertiary and quaternary protein structure prediction methods. However, due to the discrete nature of the underlying variable (amino-acids), exact inference requires exponential time in the protein length, and efficient approximations are needed for practical applicability. Here we propose a very efficient multivariate Gaussian modeling approach as a variant of direct-coupling analysis: the discrete amino-acid variables are replaced by continuous Gaussian random variables. The resulting statistical inference problem is efficiently and exactly solvable. We show that the quality of inference is comparable or superior to the one achieved by mean-field approximations to inference with discrete variables, as done by direct-coupling analysis. This is true for (i) the prediction of residue-residue contacts in proteins, and (ii) the identification of protein-protein interaction partner in bacterial signal transduction. An implementation of our multivariate Gaussian approach is available at the website http://areeweb.polito.it/ricerca/cmp/code.
Conflict of interest statement
Figures
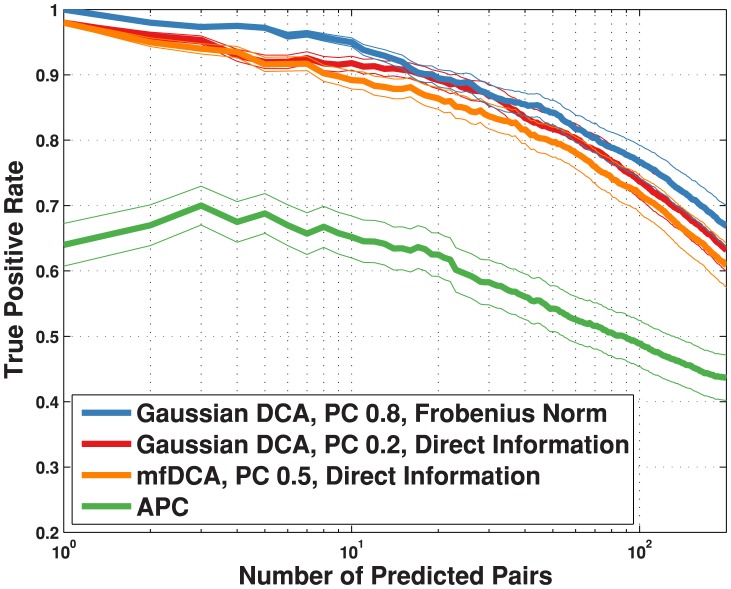
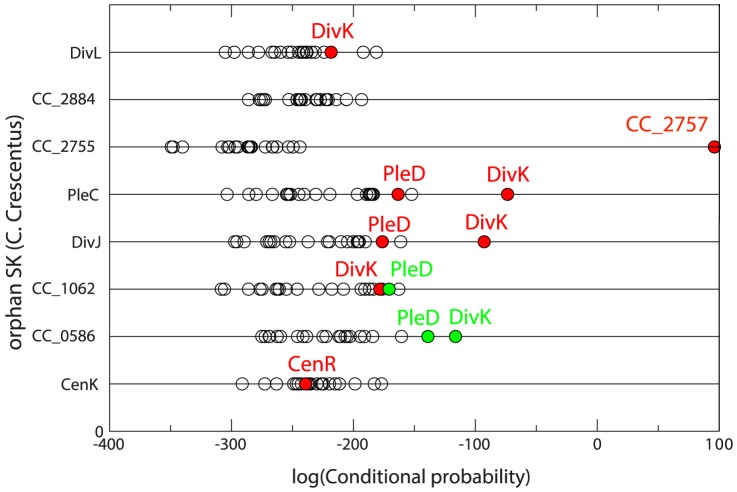
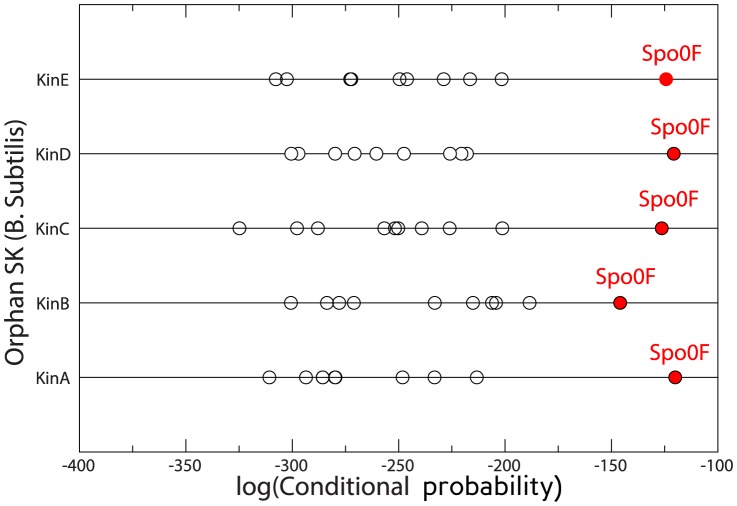
 , blue); Gaussian direct information (as described in the text, APC-corrected, pseudo-count set to
, blue); Gaussian direct information (as described in the text, APC-corrected, pseudo-count set to  , red); mean-field direct information (as described in , pseudo-count set to
, red); mean-field direct information (as described in , pseudo-count set to  , orange) and APC-corrected mutual information (as described in , green). The true positive rate is an arithmetic mean over 50 Pfam families (see Table 2 for the list); thin lines represent standard deviations.
, orange) and APC-corrected mutual information (as described in , green). The true positive rate is an arithmetic mean over 50 Pfam families (see Table 2 for the list); thin lines represent standard deviations.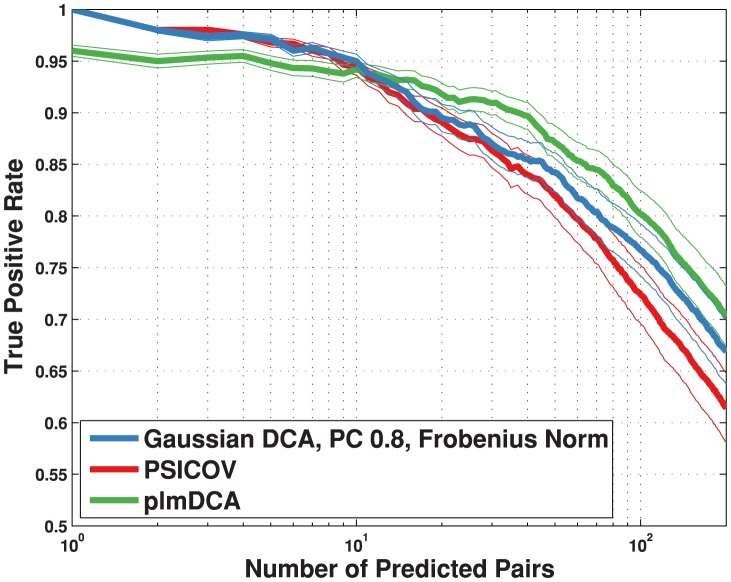
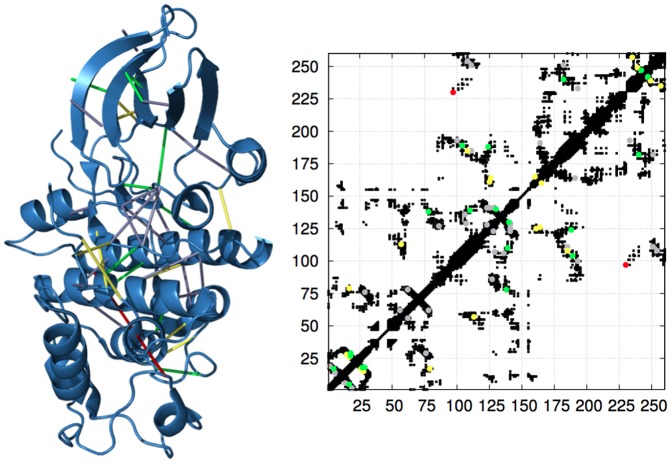
 predicted contacts are depicted in green, the next
predicted contacts are depicted in green, the next  contacts in yellow, the last
contacts in yellow, the last  contacts in grey; the only false positive contact (occurring as the 24th predicted pair) is shown in red.
contacts in grey; the only false positive contact (occurring as the 24th predicted pair) is shown in red.
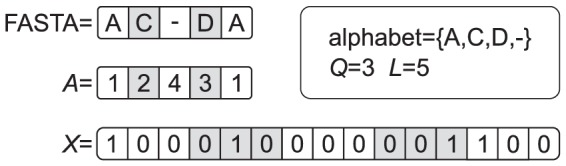
 amino-acids,
amino-acids,  , plus the gap. The alternation of white and gray cell backgrounds helps to track the transformation (e.g.
, plus the gap. The alternation of white and gray cell backgrounds helps to track the transformation (e.g.  ). Typically, MSAs of protein families are such that in every column (i.e. residue position) there appears a number of distinct residues smaller than or equal to
). Typically, MSAs of protein families are such that in every column (i.e. residue position) there appears a number of distinct residues smaller than or equal to  . Here, we did not not consider a restriction of the alphabet to the residues actually occurring, and we used instead the same encoding for all residues.
. Here, we did not not consider a restriction of the alphabet to the residues actually occurring, and we used instead the same encoding for all residues.References
-
- Altschuh D, Lesk A, Bloomer A, Klug A (1987) Correlation of co-ordinated amino acid substitutions with function in viruses related to tobacco mosaic virus. Journal of Molecular Biology 193: 693–707. - PubMed
-
- Gobel U, Sander C, Schneider R, Valencia A (1994) Correlated mutations and residue contacts in proteins. Proteins: Structure, Function and Genetics 18: 309–317. - PubMed
-
- Shindyalov I, Kolchanov N, Sander C (1994) Can three-dimensional contacts in protein structures be predicted by analysis of correlated mutations? Protein Engineering 7: 349–358. - PubMed
-
- Lockless SW, Ranganathan R (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286: 295–299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
