

The inflammatory response in myocardial injury, repair, and remodelling
- PMID: 24663091
- PMCID: PMC4407144
- DOI: 10.1038/nrcardio.2014.28
The inflammatory response in myocardial injury, repair, and remodelling
Abstract
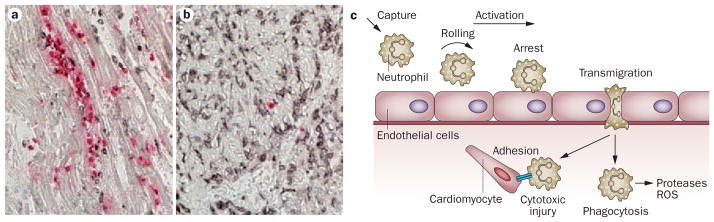
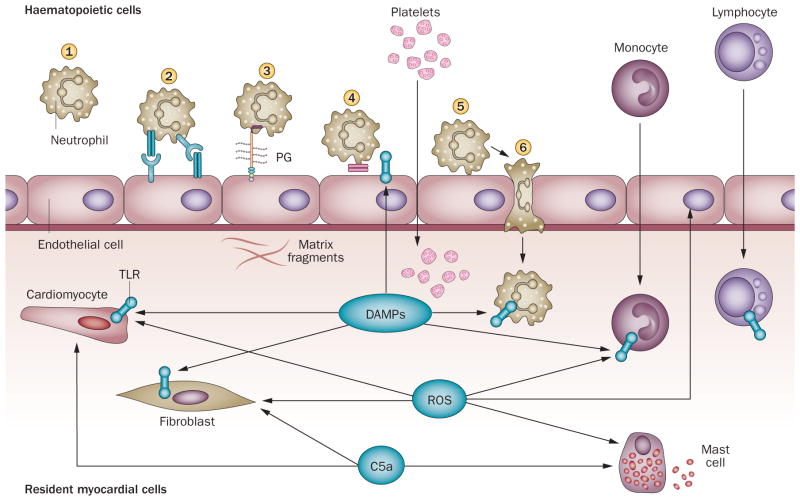
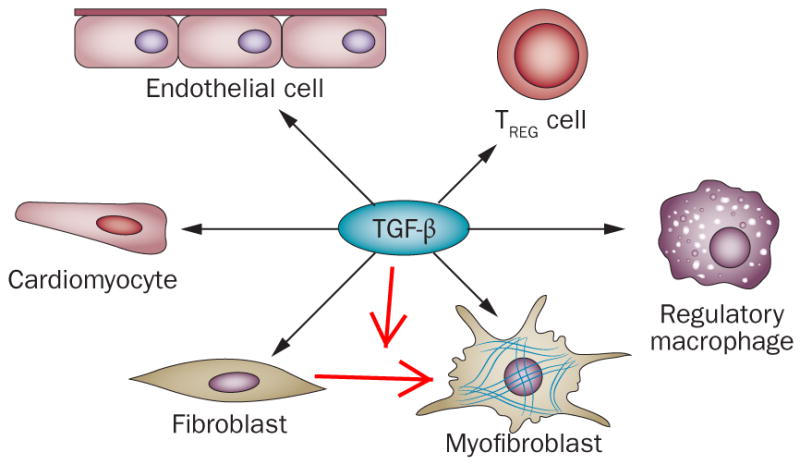
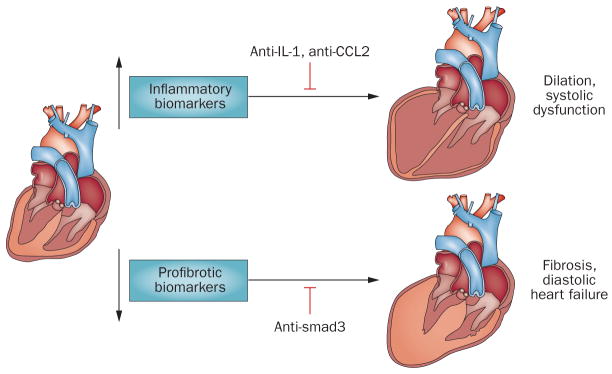
Myocardial infarction triggers an intense inflammatory response that is essential for cardiac repair, but which is also implicated in the pathogenesis of postinfarction remodelling and heart failure. Signals in the infarcted myocardium activate toll-like receptor signalling, while complement activation and generation of reactive oxygen species induce cytokine and chemokine upregulation. Leukocytes recruited to the infarcted area, remove dead cells and matrix debris by phagocytosis, while preparing the area for scar formation. Timely repression of the inflammatory response is critical for effective healing, and is followed by activation of myofibroblasts that secrete matrix proteins in the infarcted area. Members of the transforming growth factor β family are critically involved in suppression of inflammation and activation of a profibrotic programme. Translation of these concepts to the clinic requires an understanding of the pathophysiological complexity and heterogeneity of postinfarction remodelling in patients with myocardial infarction. Individuals with an overactive and prolonged postinfarction inflammatory response might exhibit left ventricular dilatation and systolic dysfunction and might benefit from targeted anti-IL-1 or anti-chemokine therapies, whereas patients with an exaggerated fibrogenic reaction can develop heart failure with preserved ejection fraction and might require inhibition of the Smad3 (mothers against decapentaplegic homolog 3) cascade. Biomarker-based approaches are needed to identify patients with distinct pathophysiologic responses and to rationally implement inflammation-modulating strategies.
Conflict of interest statement
The author declares no competing interests.
Figures
References
-
- Mallory GK, White PD, Salcedo-Salgar J. The speed of healing of myocardial infarction. A study of the pathologic anatomy in seventy-two cases. Am Heart J. 1939;18:647.
-
- Tojo SJ, et al. Reduction of rat myocardial ischemia and reperfusion injury by sialyl Lewis x oligosaccharide and anti-rat P-selectin antibodies. Glycobiology. 1996;6:463–469. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
