

Evidence of trem2 variant associated with triple risk of Alzheimer's disease
- PMID: 24663666
- PMCID: PMC3963925
- DOI: 10.1371/journal.pone.0092648
Evidence of trem2 variant associated with triple risk of Alzheimer's disease
Abstract
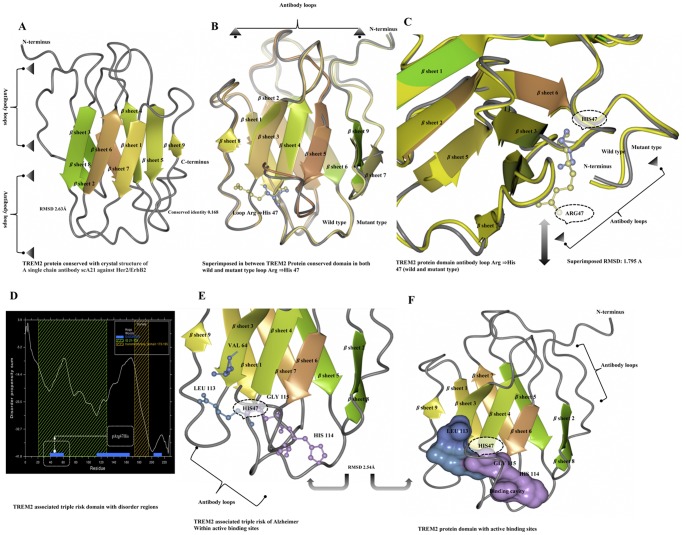
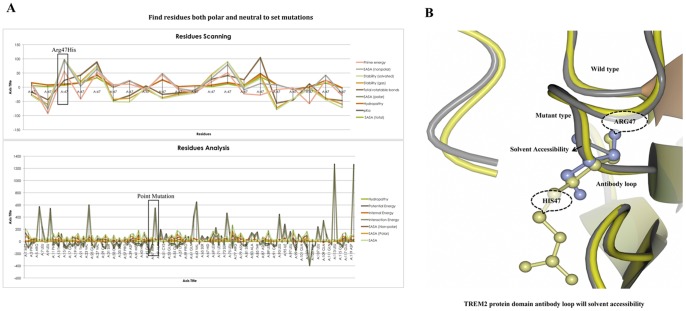
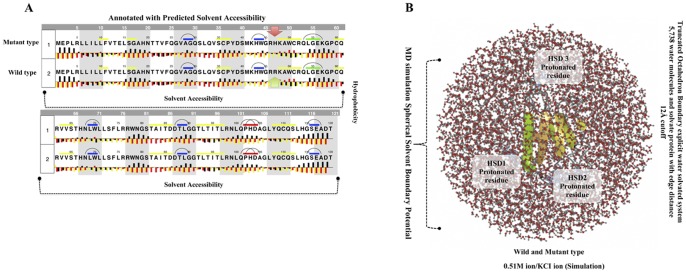
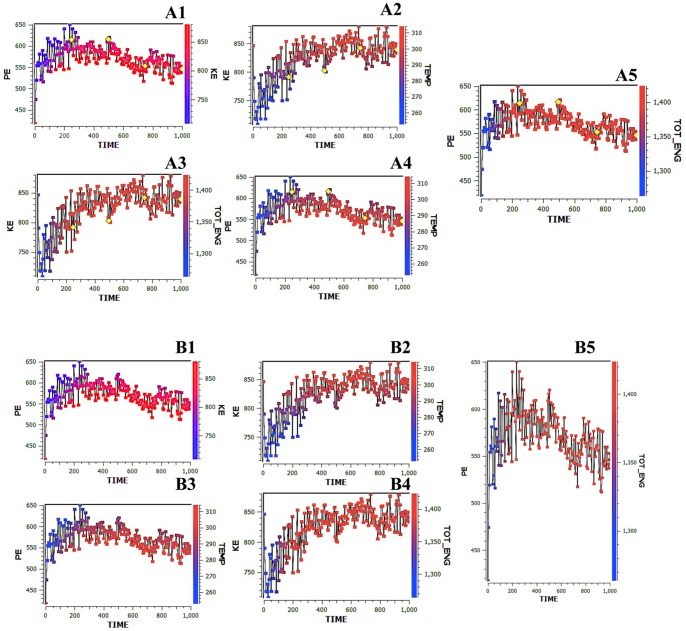
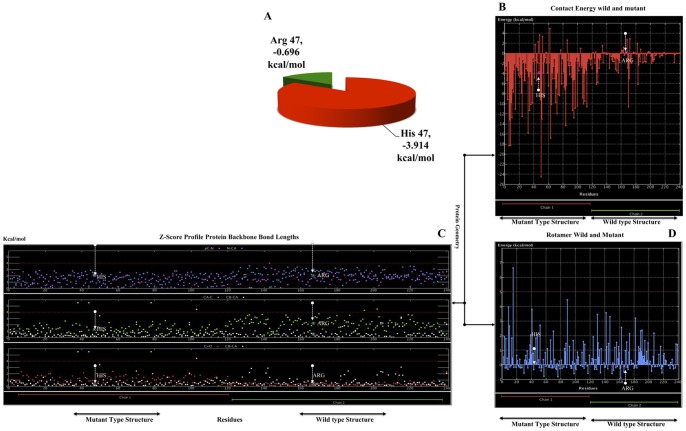
Alzheimer's disease is one of the main causes of dementia among elderly individuals and leads to the neurodegeneration of different areas of the brain, resulting in memory impairments and loss of cognitive functions. Recently, a rare variant that is associated with 3-fold higher risk of Alzheimer's disease onset has been found. The rare variant discovered is a missense mutation in the loop region of exon 2 of Trem2 (rs75932628-T, Arg47His). The aim of this study was to investigate the evidence for potential structural and functional significance of Trem2 gene variant (Arg47His) through molecular dynamics simulations. Our results showed the alteration caused due to the variant in TREM2 protein has significant effect on the ligand binding affinity as well as structural configuration. Based on molecular dynamics (MD) simulation under salvation, the results confirmed that native form of the variant (Arg47His) might be responsible for improved compactness, hence thereby improved protein folding. Protein simulation was carried out at different temperatures. At 300K, the deviation of the theoretical model of TREM2 protein increased from 2.0 Å at 10 ns. In contrast, the deviation of the Arg47His mutation was maintained at 1.2 Å until the end of the simulation (t = 10 ns), which indicated that Arg47His had reached its folded state. The mutant residue was a highly conserved region and was similar to "immunoglobulin V-set" and "immunoglobulin-like folds". Taken together, the result from this study provides a biophysical insight on how the studied variant could contribute to the genetic susceptibility to Alzheimer's disease.
Conflict of interest statement
Figures
References
-
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, et al. (2003) Protein disorder prediction: implications for structural proteomics. Structure 11: 1453–1459. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
