

Novel mutations widen the phenotypic spectrum of slow skeletal/β-cardiac myosin (MYH7) distal myopathy
- PMID: 24664454
- PMCID: PMC4112555
- DOI: 10.1002/humu.22553
Novel mutations widen the phenotypic spectrum of slow skeletal/β-cardiac myosin (MYH7) distal myopathy
Abstract
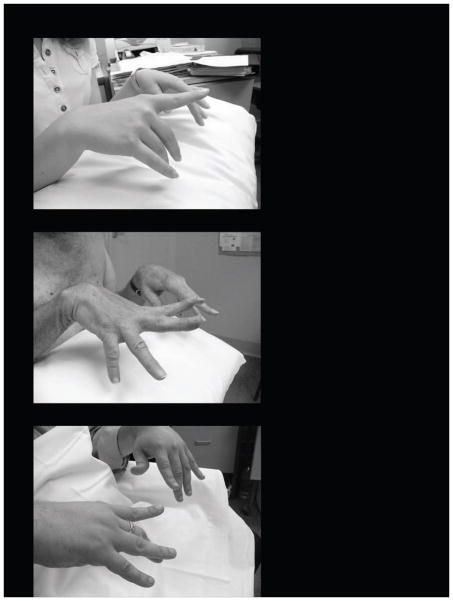
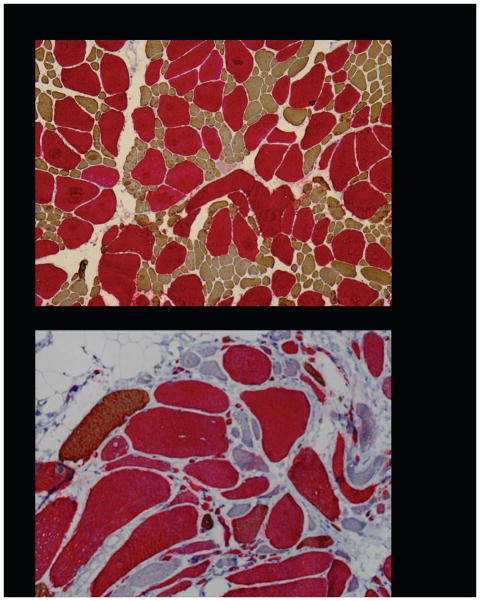
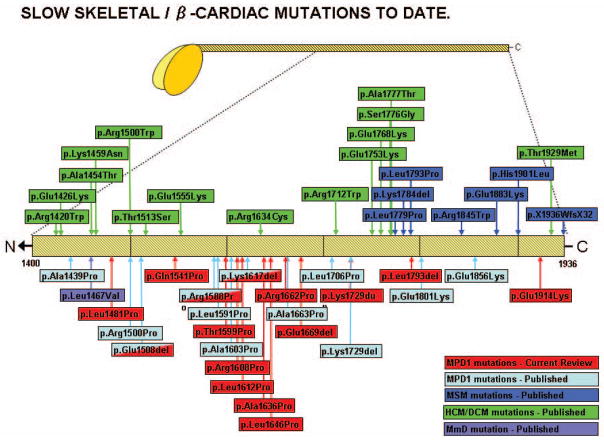
Laing early onset distal myopathy and myosin storage myopathy are caused by mutations of slow skeletal/β-cardiac myosin heavy chain encoded by the gene MYH7, as is a common form of familial hypertrophic/dilated cardiomyopathy. The mechanisms by which different phenotypes are produced by mutations in MYH7, even in the same region of the gene, are not known. To explore the clinical spectrum and pathobiology, we screened the MYH7 gene in 88 patients from 21 previously unpublished families presenting with distal or generalized skeletal muscle weakness, with or without cardiac involvement. Twelve novel mutations have been identified in thirteen families. In one of these families, the father of the proband was found to be a mosaic for the MYH7 mutation. In eight cases, de novo mutation appeared to have occurred, which was proven in four. The presenting complaint was footdrop, sometimes leading to delayed walking or tripping, in members of 17 families (81%), with other presentations including cardiomyopathy in infancy, generalized floppiness, and scoliosis. Cardiac involvement as well as skeletal muscle weakness was identified in nine of 21 families. Spinal involvement such as scoliosis or rigidity was identified in 12 (57%). This report widens the clinical and pathological phenotypes, and the genetics of MYH7 mutations leading to skeletal muscle diseases.
Keywords: Laing distal myopathy; MPD1; MYH7.
© 2014 WILEY PERIODICALS, INC.
Figures
References
-
- Borg K, Ahlberg G, Anvet M, Edstrom L. Welander distal myopathy – an overview. Neuromusc Disord. 1998;8:115–118. - PubMed
-
- Cancilla PA, Kalyanaraman K, Verity MA, Munsat T, Pearson CM. Familial myopathy with probable lysis of myofibrils in type I fibres. Neurol. 1971;21:579–585. - PubMed
-
- Chai J, Liu C, Lai P, Yee W. Myosin storage myopathy with a novel slow-skeletal myosin (MYH7) mutation in a Chinese patient. Neuromusc Disord. 2007;17:838.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
