

Diversity and dynamics of the Drosophila transcriptome
- PMID: 24670639
- PMCID: PMC4152413
- DOI: 10.1038/nature12962
Diversity and dynamics of the Drosophila transcriptome
Abstract
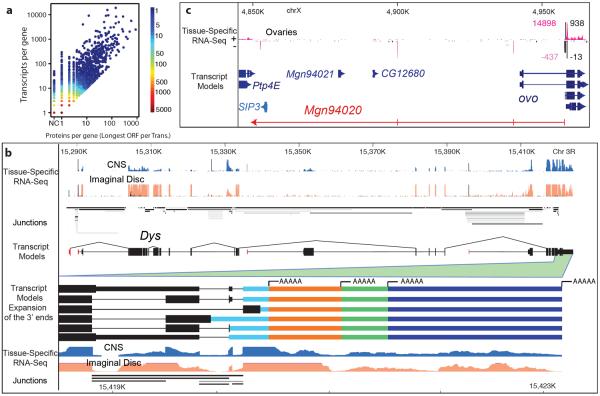
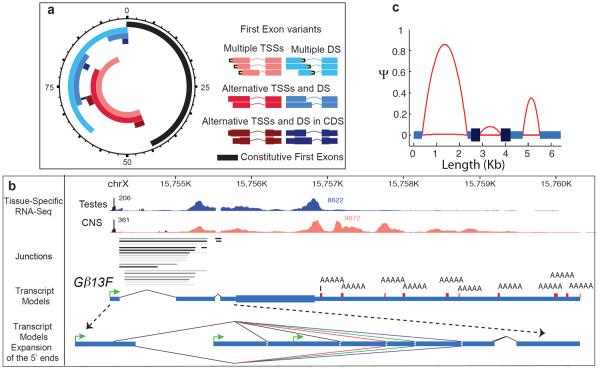
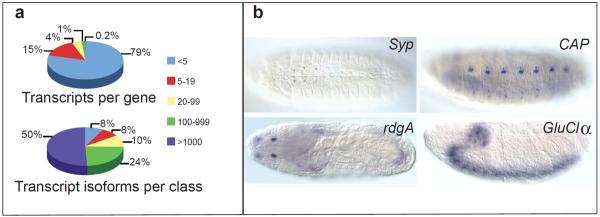
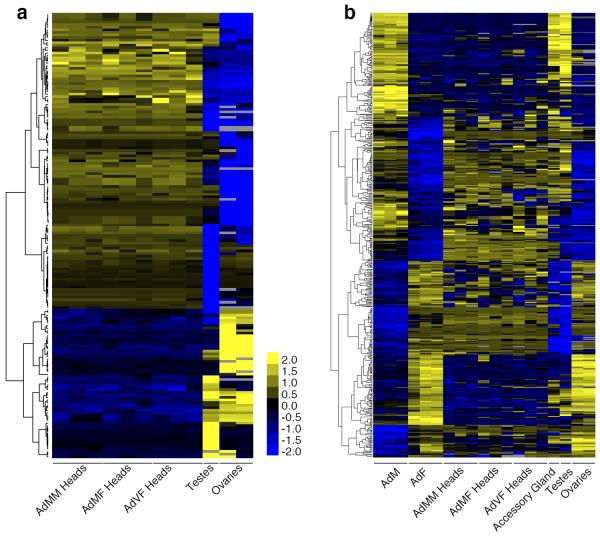
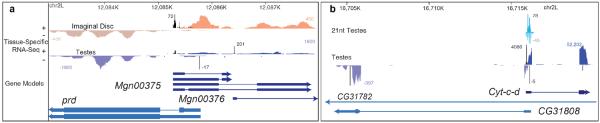
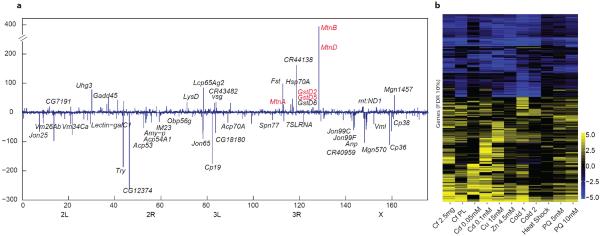
Animal transcriptomes are dynamic, with each cell type, tissue and organ system expressing an ensemble of transcript isoforms that give rise to substantial diversity. Here we have identified new genes, transcripts and proteins using poly(A)+ RNA sequencing from Drosophila melanogaster in cultured cell lines, dissected organ systems and under environmental perturbations. We found that a small set of mostly neural-specific genes has the potential to encode thousands of transcripts each through extensive alternative promoter usage and RNA splicing. The magnitudes of splicing changes are larger between tissues than between developmental stages, and most sex-specific splicing is gonad-specific. Gonads express hundreds of previously unknown coding and long non-coding RNAs (lncRNAs), some of which are antisense to protein-coding genes and produce short regulatory RNAs. Furthermore, previously identified pervasive intergenic transcription occurs primarily within newly identified introns. The fly transcriptome is substantially more complex than previously recognized, with this complexity arising from combinatorial usage of promoters, splice sites and polyadenylation sites.
Figures
Comment in
-
Discovering the complexity of the metazoan transcriptome.Genome Biol. 2014 Apr 29;15(4):112. doi: 10.1186/gb4172. Genome Biol. 2014. PMID: 25001157 Free PMC article.
-
Genomics: Hiding in plain sight.Nature. 2014 Aug 28;512(7515):374-5. doi: 10.1038/512374a. Nature. 2014. PMID: 25164742 No abstract available.
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 HG004271/HG/NHGRI NIH HHS/United States
- R01 GM076655/GM/NIGMS NIH HHS/United States
- K99 HG006698/HG/NHGRI NIH HHS/United States
- U01-HG004261/HG/NHGRI NIH HHS/United States
- RC2-HG005639/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U01 HG007031/HG/NHGRI NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- U01 HG004261/HG/NHGRI NIH HHS/United States
- U54 HG006944/HG/NHGRI NIH HHS/United States
- R01 GM097231/GM/NIGMS NIH HHS/United States
- 5U01HG004695-04/HG/NHGRI NIH HHS/United States
- 1U01HG007031-01/HG/NHGRI NIH HHS/United States
- R01 GM083300/GM/NIGMS NIH HHS/United States
- U01 HG004695/HG/NHGRI NIH HHS/United States
- RC2 HG005639/HG/NHGRI NIH HHS/United States
- ZIA DK015600/ImNIH/Intramural NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
