

High-dimensional single-cell cancer biology
- PMID: 24671264
- PMCID: PMC4216808
- DOI: 10.1007/82_2014_367
High-dimensional single-cell cancer biology
Abstract
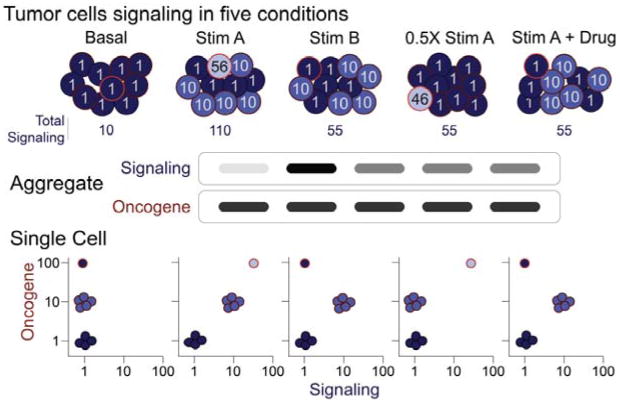
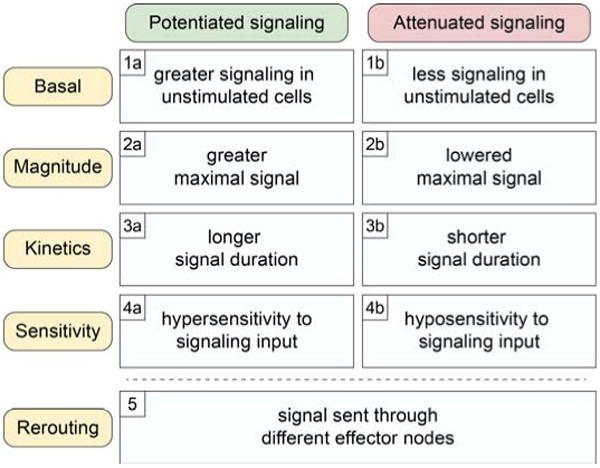
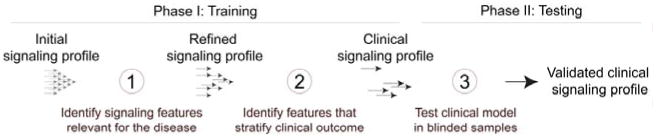
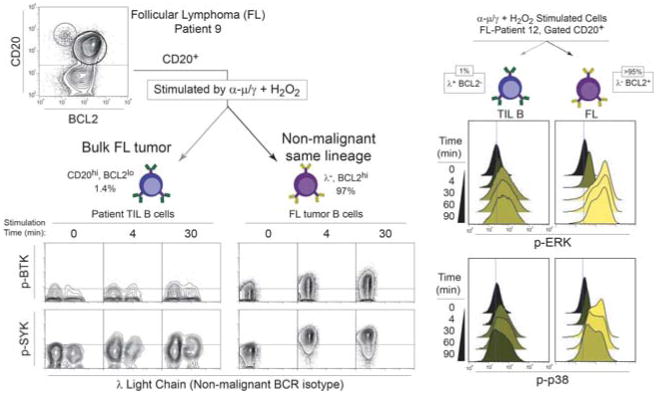
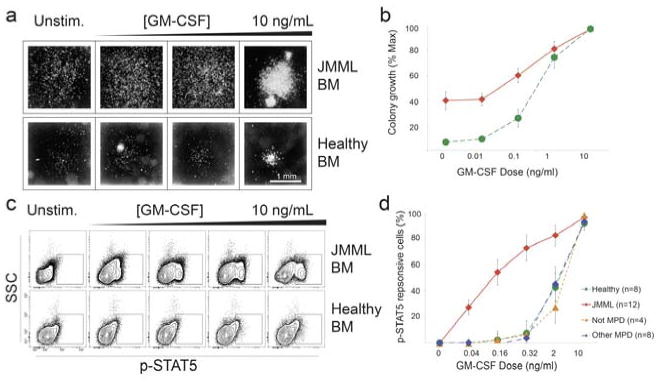
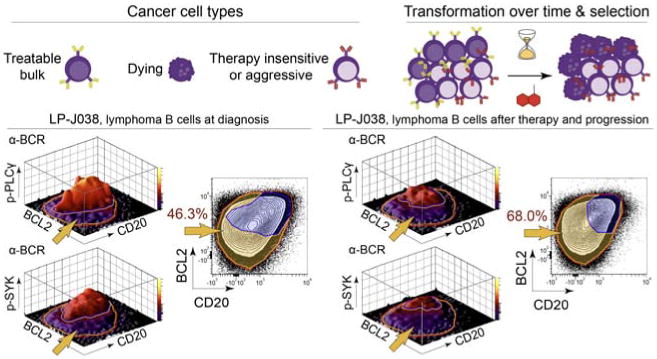
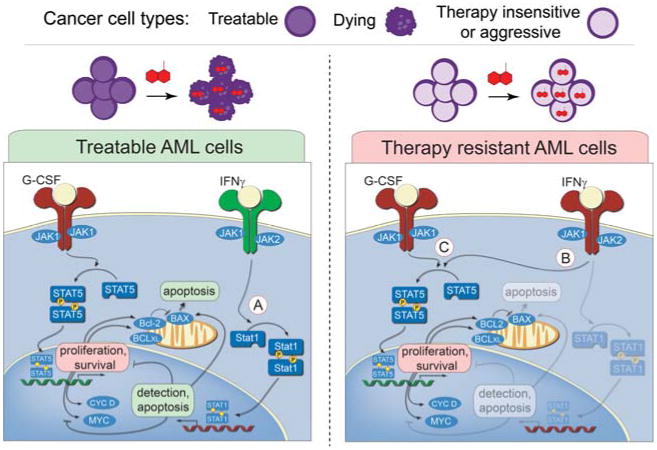
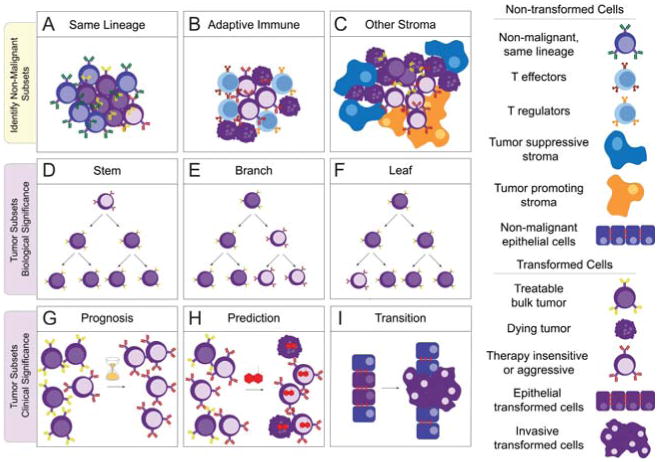
Cancer cells are distinguished from each other and from healthy cells by features that drive clonal evolution and therapy resistance. New advances in high-dimensional flow cytometry make it possible to systematically measure mechanisms of tumor initiation, progression, and therapy resistance on millions of cells from human tumors. Here we describe flow cytometry techniques that enable a "single-cell " view of cancer. High-dimensional techniques like mass cytometry enable multiplexed single-cell analysis of cell identity, clinical biomarkers, signaling network phospho-proteins, transcription factors, and functional readouts of proliferation, cell cycle status, and apoptosis. This capability pairs well with a signaling profiles approach that dissects mechanism by systematically perturbing and measuring many nodes in a signaling network. Single-cell approaches enable study of cellular heterogeneity of primary tissues and turn cell subsets into experimental controls or opportunities for new discovery. Rare populations of stem cells or therapy-resistant cancer cells can be identified and compared to other types of cells within the same sample. In the long term, these techniques will enable tracking of minimal residual disease (MRD) and disease progression. By better understanding biological systems that control development and cell-cell interactions in healthy and diseased contexts, we can learn to program cells to become therapeutic agents or target malignant signaling events to specifically kill cancer cells. Single-cell approaches that provide deep insight into cell signaling and fate decisions will be critical to optimizing the next generation of cancer treatments combining targeted approaches and immunotherapy.
Figures
References
-
- Andreeff M, Slater DE, Bressler J, Furth ME. Cellular ras oncogene expression and cell cycle measured by flow cytometry in hematopoietic cell lines. Blood. 1986;67:676–81. - PubMed
-
- Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, et al. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002;9:252–63. - PubMed
-
- Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1:2365–76. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
